COG STIM2
Stimulation cognitive informatisée supervisée à distance visant à réduire les difficultés cognitives après chimiothérapie chez les femmes traitées pour un cancer du sein localisé : étude contrôlée randomisée multicentrique
Type d'essai : Académique / Institutionnel
Etat de l'essai : Ouvert
Situation thérapeutique : Tumeur solide ( Adjuvant )
Etablissement(s) participant(s)
Dr Flora COURTAULT-DESLANDES
Dr Nathalie PINTO
Dr Lorène SEGUIN
Détails de l'essai
Objectif principal
Evaluer, chez des patientes atteintes d’un cancer du sein localisé, le bénéfice d’un programme de stimulation cognitive informatisé de 12 semaines supervisé par un neuropsychologue (groupe expérimental) sur les plaintes cognitives par rapport à un accès libre non supervisé de 12 semaines au même programme (groupe témoin).
L’évaluation du bénéfice sera basée sur le changement des plaintes cognitives à la fin du programme par rapport à la baseline.
Objectif(s) secondaire(s)
Evaluer et comparer deux groupes de patientes atteintes d’un cancer du sein en fonction de plusieurs paramètres, notamment :
– L’adhésion individuelle des patientes au programme de stimulation cognitive en ligne dans chaque groupe.
– Changement dans les plaintes cognitives à T1 (fin de l’intervention), T2 (3 mois après l’intervention) et T3 (9 mois après l’intervention).
– Changement dans les performances cognitives objectives à T1, T2 et T3.
– Qualité de vie liée à la santé des patients à T1, T2 et T3.
– Niveaux d’activité physique à T1, T2 et T3.
– Changements de la fatigue, du sommeil, de l’anxiété et de la dépression des patients à T1, T2 et T3.
– La relation entre la fatigue, le sommeil, l’anxiété, la dépression, l’activité physique et les plaintes/performances cognitives.
– La proportion de patients qui retournent au travail à T1, T2 et T3 parmi les patients qui travaillent.
– L’impact médico-économique de l’intervention.
– L’évolution des paramètres biologiques.
Résumé / schéma de l'étude
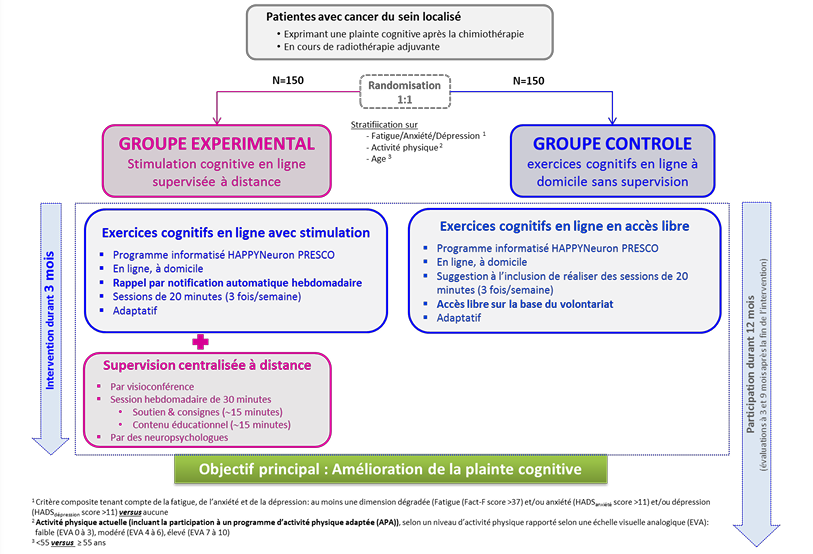
Critère(s) d'inclusion
- Patiente présentant un diagnostic de cancer du sein localisé.
- Agée de 18 ans ou plus.
- Qui a reçu une chimiothérapie adjuvante ou néo-adjuvante et qui reçoit actuellement une radiothérapie adjuvante (une hormonothérapie continue est autorisée) jusqu’à 30 jours après la fin de la radiothérapie.
- Patiente rapportant des plaintes cognitives ayant un impact significatif sur leur qualité de vie, telles qu’évaluées par la sous-échelle de qualité de vie du questionnaire FACT-Cog. Cette sous-échelle est composée de 4 questions :
1. J’ai été bouleversé par ces problèmes.
2. Ces problèmes ont interféré avec ma capacité à travailler.
3. Ces problèmes m’ont empêché de faire des choses que j’aime.
4. Ces problèmes ont interféré avec la qualité de ma vie.
Les patientes seront éligibles si leur score à cette sous-échelle est égal à 2 ou plus.
Les patientes sont éligibles si leur score à cette sous-échelle est égal ou inférieur au 10ème percentile, sur la base des directives relatives à l’âge et des données normatives (Lange et al., 2015), à savoir :- ≤ 8 pour les patientes âgées de 30 à 49 ans.
- ≤ 9 pour les patientes âgées de 50 à 69 ans.
- ≤ 10 pour les patientes âgées de 70 à 89 ans.
- Patiente ayant suivi au moins trois années d’enseignement primaire, selon l’échelle de Barbizet.
- Patiente ayant accès à un ordinateur portable fonctionnel doté d’un clavier, d’une connexion internet et d’un compte de messagerie électronique, et capable d’utiliser ces outils seule.
- Patiente parlant couramment le français.
- Patiente ayant donné leur consentement éclairé de participation à l’étude.
Critère(s) de non-inclusion
- Trouble de la personnalité ou toute pathologie psychiatrique évolutive connue (par exemple, schizophrénie).
- Antécédents neurologiques avec symptômes cognitifs persistants (séquelles de traumatisme crânien, accident vasculaire cérébral, sclérose en plaques, épilepsie, pathologie neurodégénérative, etc,).
- Consommation excessive d’alcool ou de drogues, qui pourrait compromettre la participation à l’intervention.
- Déficit visuel et/ou auditif important.
- Patiente qui pourrait ne pas être en mesure de compléter les tests neuropsychologiques, (y compris ceux avec des troubles cognitifs significatifs qui empêchent la réalisation des tests cognitifs, tel que déterminé par le test de dépistage cognitif MoCA et basé sur l’âge et le niveau d’éducation selon les données normatives du GRECOGVASC).
- Participation actuelle à un programme d’entraînement cognitif.
- Refus de participer.
- Patiente privée de liberté ou sous tutelle.
- Patiente qui pourrait ne pas être en mesure de participer pour des raisons géographiques, sociales ou psychopathologiques.
Calendrier prévisionnel
Lancement de l’étude : Octobre 2024
Fin estimée des inclusions : Octobre 2028
Nombre de patients à inclure : 300
Coordonnateur de l'étude
Pr Florence JOLY – Centre François Baclesse – CLCC CAEN
Promoteur de l'étude
Centre François Baclesse – CLCC Caen