MorningLyte
Etude de Phase III, randomisée, ouverte, internationale, mutlicentrique, évaluant l’efficacité et la tolérance du Mosunetuzumab associé au Lenalidomide anticorps monoclonal anti CD20 associé à une chimiothérapie chez des patients atteints d’un lymphome folliculaire FLIPI 2-5 non préalablement traité
Type d'essai : Académique / Institutionnel
Etat de l'essai : Ouvert
Situation thérapeutique : Hématologie ( De Novo )
Etablissement(s) participant(s)
Dr Gabriel BRISOU
Dr Hacène ZERAZHI
Détails de l'essai
Objectif principal
Survie sans progression (PFS).
Résumé / schéma de l'étude
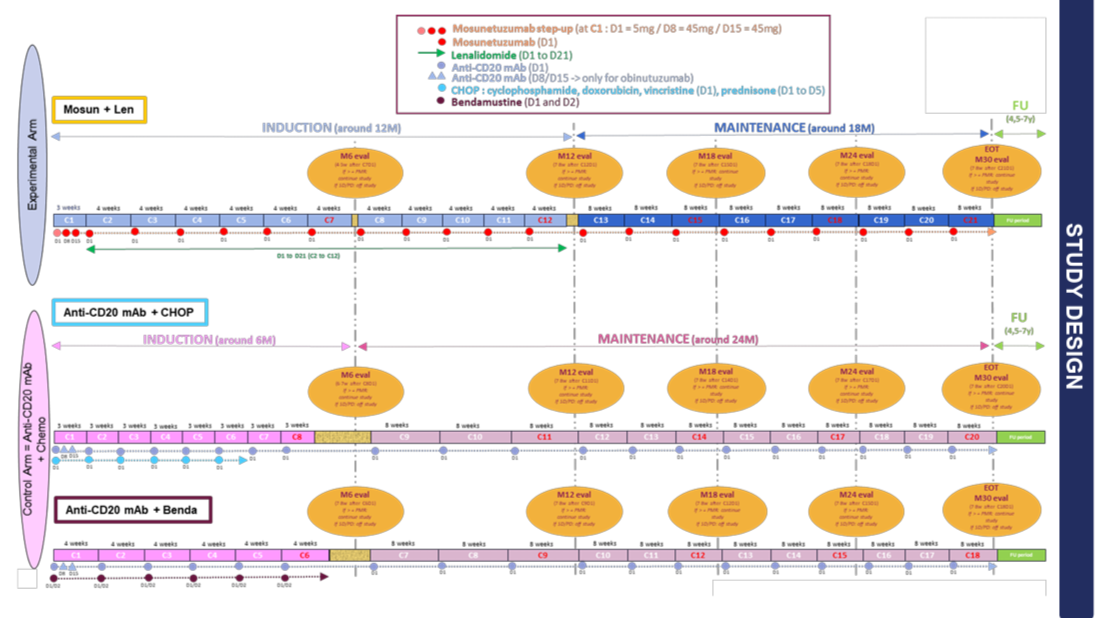
Expérimental : Mosun-Len
Dans le bras expérimental, les patients seront traités pendant 1 cycle de 3 semaines pour le mosunetuzumab puis 11 cycles de 4 semaines pour le mosunetuzumab et le lénalidomide (47 semaines, environ 11 mois) pendant la phase d’induction, et pendant un maximum de 9 cycles supplémentaires de 8 semaines pendant la phase d’entretien (72 semaines, environ 17 mois), jusqu’à environ 125 semaines (30 mois). Les patients doivent commencer la phase d’entretien 7 à 8 semaines après le début du dernier cycle d’induction (C12).
Comparateur actif :
Dans le bras témoin, les patients seront traités pendant 8 ou 6 cycles de 3 ou 4 semaines pour un mAb anti-CD20 + cyclophosphamide-doxorubicine-vincristine-prednisone (CHOP) ou un mAb anti-CD20 + Bendamustine, respectivement, selon le bras attribué. (24 semaines, environ 5 mois) pendant la phase d’induction, et pendant un maximum de 12 cycles supplémentaires de 8 semaines pendant la phase d’entretien (96 semaines, environ 22 mois), jusqu’à environ 125 semaines (30 mois). Les patients doivent commencer la phase d’entretien 6 à 7 ou 7 à 8 semaines après le début du dernier cycle d’induction (C8 ou C6).
L’option de passage du bras de contrôle au bras expérimental n’est pas autorisée.
Tous les patients randomisés seront suivis pour une survie sans progression et une survie globale en utilisant le même calendrier. Les patients seront suivis depuis l’évaluation de fin de traitement tous les 3 mois pendant les deux premières années, puis tous les 6 mois pendant les 3 années suivantes, puis annuellement jusqu’à la fin de l’étude.
La fin de l’étude surviendra lorsque tous les patients randomisés auront été suivis pour leur survie pendant au moins 7 ans (ou auront arrêté l’étude prématurément).
Critère(s) d'inclusion
- Patient atteint d’un lymphome folliculaire CD20+ non traité préalablement prouvé histologiquement, de grade 1, 2 ou 3a (y compris le patient suivi pendant jusqu’à 10 ans après le diagnostic initial) tel qu’évalué par les enquêteurs selon la classification 2016 de l’Organisation mondiale de la santé (OMS)12, ou lymphome folliculaire classique selon la classification OMS 202213. Les tissus diagnostiques doivent être disponibles pour l’examen central de la pathologie, les critères d’évaluation exploratoires et l’utilisation des données secondaires. (Les patients avec un nombre absolu de lymphocytes > 20 G/L doivent être discutés avec le promoteur avant le dépistage/l’inclusion).
- FLIP 2-5.
- Tous les stades d’Ann Arbor (y compris le stade I si FLIPI ≥ 2).
- Doit nécessiter un traitement comme en témoigne au moins un des critères suivants :
- Maladie volumineuse définie comme :
- Masse/lésion ganglionnaire ou extraganglionnaire > 7 cm dans son plus grand diamètre ou,
- Atteinte d’au moins 3 sites nodaux ou extraganglionnaires (chacun avec un diamètre supérieur à > 3 cm
- Présence d’au moins un des symptômes B suivants :
- Fièvre (> 38°C) d’étiologie incertaine
- Sueurs nocturnes
- Perte de poids supérieure à 10 % au cours des 6 mois précédents
- Splénomégalie symptomatique
- Tout syndrome compressif (par exemple, mais sans s’y limiter : urétéral, orbitaire, gastro-intestinal)
- L’une des cytopénies suivantes dues à un lymphome :
-Hémoglobine < 10 g/dL (6,25 mmol/L)
-Plaquettes <100 x 109/L, ou
-Nombre absolu de neutrophiles (ANC) < 1,5 x 109/L
-Epanchement séreux pleural ou péritonéal (quel que soit le contenu cellulaire)
-β2microglobuline > LSN ou lactate déshydrogénase (LDH) > LSN
- Au moins une lésion ganglionnaire bidimensionnellement mesurable, définie comme > 1,5 cm dans sa dimension la plus longue, ou au moins une lésion ganglionnaire extra-ganglionnaire bidimensionnellement mesurable, définie comme > 1,0 cm dans sa dimension la plus longue (et 18F-2-fluoro-2 -désoxy-D-glucose (FDG)-lésion avide).
- Patient qui a compris et volontairement signé et daté un consentement éclairé avant toute évaluation/procédure spécifique à l’étude.
- Doit être ≥ 18 ans au moment de la signature du formulaire de consentement éclairé (ICF).
- Statut de performance de l’Eastern Cooperative Oncology Group (ECOG) 0 à 2.
- Espérance de vie minimale estimée à 3 mois.
- Fonction hématologique adéquate dans les 28 jours précédant la signature du consentement éclairé, notamment :
- Nombre absolu de neutrophiles (ANC) ≥ 1 x 109/L
- Numération plaquettaire ≥ 75 x 109/L, ou ≥ 30 x 109/L en cas d’infiltration médullaire ou de splénomégalie
- Hémoglobine ≥ 8,0 g/dL (5 mmol/L) sauf en cas d’infiltration médullaire ou de splénomégalie. La transfusion est autorisée avant le début du traitement (aucune fenêtre requise)
- Valeurs normales de laboratoire :
- Clairance de la créatinine mesurée ou estimée ≥ 40 mL/min calculée selon la méthode standard de l’établissement (MDRD ou Cockcroft-Gault)
- Aspartate aminotransférase (AST) ou alanine aminotransférase (ALT) ≤ 2,5 x LSN, sauf chez les patients présentant une atteinte hépatique ou pancréatique documentée par un lymphome ≤ 5 x LSN
- Bilirubine totale sérique ≤ 1,5 x LSN (ou ≤ 3 x LSN pour les patients atteints du syndrome de Gilbert), sauf chez les patients présentant une atteinte hépatique ou pancréatique documentée par lymphome ≤ 3 x LSN
- Fraction d’éjection ventriculaire gauche (FEVG) dans les limites normales (c’est-à-dire > 50 % tel qu’évalué par échocardiographie transthoracique ou > 45 % tel qu’évalué par méthode isotopique (scan MUGA)).
- Les patients doivent pouvoir recevoir une prophylaxie et/ou un traitement adéquat en cas d’événements thromboemboliques (aspirine, héparine de bas poids moléculaire ou anticoagulants oraux directs).Les patients bénéficiant d’un traitement anticoagulant curatif peuvent être inscrits. Un patient présentant une thrombose veineuse profonde due à un syndrome compressif est éligible si un traitement anticoagulant curatif a été débuté au moins 1 semaine avant le début du traitement à l’étude : héparine de bas poids moléculaire possible au début du traitement, puis anticoagulants oraux directs selon les pratiques locales.
- Doit être en mesure de respecter le calendrier des visites d’étude et les autres exigences du protocole.
- Test SARS-CoV-2 négatif dans les 7 jours précédant la randomisation. Un test antigénique rapide est également acceptable. Si un patient a un test SARS-CoV-2 positif avant la randomisation, un autre test doit être effectué et être négatif dans les 7 jours précédant le début du traitement.
- Test VIH négatif avant randomisation, à l’exception suivante : les patients dont le test VIH est positif avant la randomisation sont éligibles à condition qu’ils soient stables sous traitement antirétroviral depuis au moins 4 semaines, qu’ils aient un taux de CD4 ≥ 200/µL, qu’ils aient une charge virale indétectable et qu’ils n’aient pas d’antécédents d’infection opportuniste attribuable au SIDA. au cours des 12 derniers mois.
- Pour les femmes en âge de procréer (WOCBP) (se référer à la section 14.7) : doivent avoir un résultat négatif au test de grossesse (sérum hautement sensible) lors du dépistage et dans les 7 jours précédant le début du traitement à l’étude, et accepter de s’abstenir d’allaiter pendant la participation à l’étude, et pendant au moins 28 jours après la dernière dose de lénalidomide (le cas échéant), 3 mois après la dernière dose de mosunetuzumab et de tocilizumab (le cas échéant), 6 mois après la dernière dose de chimiothérapies (le cas échéant), 12 mois après la dernière dose de chimiothérapie. dose de rituximab (le cas échéant) et 18 mois après la dernière dose d’obinutuzumab (le cas échéant).
- Pour les hommes (se référer à la section 14.7) : Accord à rester abstinent (s’abstenir de rapports hétérosexuels) ou à utiliser un préservatif, et accord à s’abstenir de donner du sperme, tel que défini ci-dessous : avec une partenaire féminine en âge de procréer ou une partenaire enceinte, les hommes doivent rester abstinent ou utiliser un préservatif pendant la période de traitement (y compris les périodes d’interruption du traitement) et pendant au moins 28 jours après la dernière dose de lénalidomide (le cas échéant), 2 mois après la dernière dose de tocilizumab (le cas échéant), 6 mois après la dernière dose de chimiothérapies (le cas échéant), 12 mois après la dernière dose de rituximab (le cas échéant) et 3 mois après la dernière dose d’obinutuzumab (le cas échéant).
- Patient couvert par tout système de sécurité sociale (France).
- Patient qui comprend et parle l’une des langues officielles du pays, sauf si la réglementation locale autorise les traducteurs indépendants.
Critère(s) de non-inclusion
- Lymphome folliculaire de grade 3b selon la classification OMS 201612, ou lymphome folliculaire à grandes cellules B selon la classification OMS 202213.
- Suspicion ou preuve clinique d’un lymphome transformé lors du recrutement par l’évaluation de l’investigateur (par exemple, SUV élevée dans au moins une lésion qui n’a pas été biopsiée et discordante avec la SUV de la lésion biopsiée (au moins le double de la SUV moyenne), LDH > 2,5 x LSN en particulier dans un contexte de maladie à évolution rapide, etc. (Veuillez contacter le Promoteur pour discuter de toute inclusion possible dans les cas limites ou en cas de doute).
- Radiothérapie localisée antérieure pour le FL.
- Antécédents d’un autre lymphome.
- Epanchement pleural ou séreux symptomatique incontrôlé nécessitant un traitement urgent dans les 48 heures (les patients dont la maladie est contrôlée après un drainage pleural/séreux adéquat et/ou un pleurX™ efficace ou un système similaire sont éligibles).
- Uretérohydronéphrose symptomatique non contrôlée entraînant une insuffisance rénale (les patients bénéficiant d’une prise en charge adéquate, c’est-à-dire un cathéter urétéral ou un stent double J permettant le contrôle de l’insuffisance rénale sont éligibles).
- Lésion épidurale lymphomateuse symptomatique (les patients dont la maladie est contrôlée par neurochirurgie ou une courte cure de stéroïdes sont éligibles).
- Utilisation de tout traitement médicamenteux anticancéreux standard ou expérimental dans les 42 jours suivant le début (jour 1) du traitement à l’étude.
- Médicaments immunosuppresseurs systémiques (y compris, mais sans s’y limiter, le cyclophosphamide, l’azathioprine, le méthotrexate, la thalidomide et les agents anti-facteur de nécrose tumorale) ou corticostéroïde > 1 mg/kg/jour de prednisone ou équivalent dans les 10 jours précédant la première dose du traitement à l’étude. Un traitement systémique par corticostéroïdes < 20 mg/jour de prednisone ou équivalent, des corticostéroïdes inhalés et des minéralocorticoïdes pour la prise en charge de l’hypotension orthostatique est autorisé. Une dose unique de dexaméthasone pour les nausées ou les symptômes B est autorisée.
- A reçu un vaccin vivant atténué dans les 4 semaines précédant la première dose du traitement à l’étude, ou chez qui il est prévu qu’un tel vaccin vivant atténué sera nécessaire pendant la période d’étude ou dans les 5 mois après la dernière dose du traitement à l’étude.
- Chirurgie majeure (à l’exclusion de la documentation chirurgicale du FL) dans les 28 jours précédant la signature du consentement éclairé.
- Séropositif ou infection virale active par le virus de l’hépatite B (VHB) :
- AgHBs positif
- AgHBs négatif, anti-HBs positif et/ou anti-HBc positif et ADN viral détectable (les patients qui sont AgHBs négatifs, anti-HBs positifs et/ou anti-HBc positifs mais ADN viral négatif sont éligibles). Ils doivent être traités et effectuer des tests à intervalles réguliers ; Les patients séropositifs en raison d’antécédents de vaccin contre l’hépatite B (anti-HBs positifs) sont éligibles).
- Séropositif connu ou infecté activement par le virus de l’hépatite C (VHC) (les patients qui sont positifs pour les anticorps anti-VHC avec un ARN viral négatif sont éligibles).
- Hypersensibilité connue ou suspectée aux produits biopharmaceutiques produits dans les cellules ovariennes de hamster chinois (CHO) ou à tout composant du mosunetuzumab, de l’AcM anti-CD20, du tocilizumab, de la formulation de lénalidomide, y compris le mannitol ; ou à l’un des excipients.
- Antécédents de transplantation d’organe solide ou de greffe allogénique de cellules souches (SCT).
- Maladie auto-immune active nécessitant un traitement.
- Antécédents de maladie auto-immune, y compris, mais sans s’y limiter, myasthénie grave, myosite, hépatite auto-immune, lupus érythémateux disséminé, polyarthrite rhumatoïde, maladie inflammatoire de l’intestin, thrombose vasculaire associée au syndrome des antiphospholipides, granulomatose de Wegener, syndrome de Sjögren, syndrome de Guillain-Barré, syndromes multiples sclérose, vascularite ou glomérulonéphrite.
- Les participants ayant des antécédents d’hypothyroïdie auto-immune et recevant une dose stable d’hormone thyroïdienne de remplacement peuvent être éligibles.
- Les participants atteints de diabète sucré de type 1 contrôlé qui suivent un régime d’insuline sont éligibles pour l’étude.
- Les participants ayant des antécédents de purpura thrombocytopénique immunitaire lié à une maladie ou d’anémie hémolytique auto-immune peuvent être éligibles.
- Les participants ayant des antécédents lointains ou une maladie auto-immune bien contrôlée, avec un intervalle sans traitement depuis le traitement immunosuppresseur pendant 12 mois peuvent être éligibles après examen et discussion avec l’investigateur principal.
- Participants présentant une infection active telle qu’une infection bactérienne, virale (y compris le SRAS-CoV-2), fongique, mycobactérienne, parasitaire ou autre (à l’exclusion des infections fongiques des lits d’ongles) active connue ou suspectée, un virus Epstein-Barr (EBV) actif chronique connu ou suspecté. ) infection, sont exclus.
- Preuve de toute maladie concomitante importante qui pourrait affecter le respect du protocole ou l’interprétation des résultats, y compris, mais sans s’y limiter :
- maladie cardiovasculaire importante [par exemple, maladies cardiaques de classe objective C ou D (cf. Classes d’insuffisance cardiaque | American Heart Association), infarctus du myocarde au cours des 6 mois précédents, arythmie instable ou angine instable)
- maladie pulmonaire importante (telle qu’une maladie pulmonaire obstructive ou des antécédents de bronchospasme)
- antécédents cliniquement significatifs de maladie du foie, y compris une hépatite virale ou autre, ou une cirrhose
- antécédents actuels ou passés de maladie du système nerveux central (SNC), comme un accident vasculaire cérébral, l’épilepsie, une vascularite du SNC ou une maladie neurodégénérative. Les participants ayant des antécédents d’accident vasculaire cérébral qui n’ont pas subi d’accident vasculaire cérébral ou d’accident ischémique transitoire au cours de la dernière année et n’ont aucun déficit neurologique résiduel jugé par l’investigateur sont autorisés. Les participants ayant des antécédents d’épilepsie et n’ayant eu aucune crise au cours des 2 dernières années avec ou sans médicaments antiépileptiques peuvent être éligibles.
- Antécédents de leucoencéphalopathie multifocale progressive confirmée (LEMP).
- Antécédents connus ou suspectés de lymphohistiocytose hémophagocytaire (HLH).
- Antécédents d’érythème polymorphe, d’éruption cutanée de grade ≥ 3 ou d’éruption cutanée vésculante après un traitement antérieur par des dérivés immunomodulateurs.
- Antécédents de maladie pulmonaire interstitielle (MPI), de pneumopathie d’origine médicamenteuse et de pneumopathie auto-immune.
- Malignité active autre que celle traitée dans cette recherche. Antécédents de tumeurs malignes, sauf si le patient est indemne de la maladie depuis ≥ 3 ans. Cependant, les patients présentant les antécédents/conditions concomitantes suivants sont éligibles :
- Cancer de la peau localisé sans mélanome.
- Carcinome in situ du col de l’utérus.
- Carcinome in situ du sein.
- Découverte histologique fortuite d’un cancer de la prostate (T1a ou T1b selon le système de stadification des métastases ganglionnaires tumorales (TNM)) ou d’un cancer de la prostate qui a été traité à visée curative.
- Présence ou antécédents d’atteinte du SNC ou méningée par lymphome.
- Enceinte, envisageant de le devenir ou allaitante WOCBP.
- Toute condition médicale significative, y compris la présence d’une anomalie de laboratoire ou d’une maladie psychiatrique qui expose le patient à un risque inacceptable s’il devait participer à l’étude, et susceptible d’interférer avec la participation à cette étude clinique (selon la décision de l’investigateur) ou ce qui confond la capacité à interpréter les données de l’étude.
- Personne privée de sa liberté par une décision judiciaire ou administrative.
- Personne hospitalisée sans consentement.
- Personne majeure sous protection légale.
Nota bene : depuis le 28., s’il y a un bénéfice individuel pour ces patients, un Comité d’Éthique devra en être informé au cas par cas.
Calendrier prévisionnel
Lancement de l’étude : Juillet 2024
Fin estimée des inclusions : Novembre 2028
Nombre de patients à inclure : 790
Coordonnateur de l'étude
Pr Franck MORSCHHAUSER – CHU Lille
Promoteur de l'étude
Lymphoma Academic Research Organisation (LYSARC)