ROCSAN (2ème partie)
Etude de phase II, randomisée, internationale, multicentrique évaluant le dostarlimab associé au niraparib versus le niraparib seul comparé à la chimiothérapie chez des patientes atteintes d’un carcinosarcome métastasique ou en récidive de l’ovaire ou de l’endomètre après une chimiothérapie de première intention
Type d'essai : Académique / Institutionnel
Etat de l'essai : Ouvert
Situation thérapeutique : Tumeur solide ( Localement avancée / Non résécable , Métastatique / Rechute )
Etablissement(s) participant(s)
Dr Magali PROVENSAL
Détails de l'essai
Objectif principal
Phase II – Etape 1, phase de sélection :
Sélectionner la meilleure stratégie thérapeutique entre l’association niraparib et dostarlimab, et le niraparib en monothérapie.
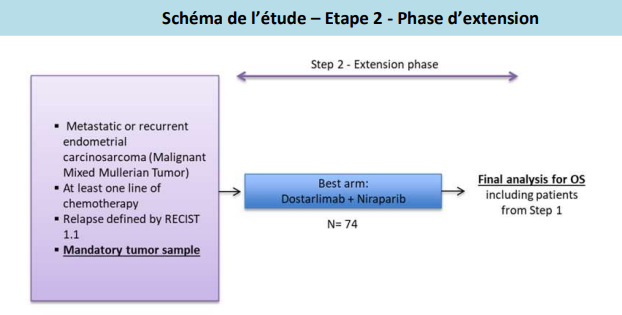
Phase II – Etape 2, phase d’extension :
Evaluer l’efficacité de la meilleure stratégie expérimentale chez les patientes ayant un carcinosarcome en progression ou récidivant de l’endomètre, et ayant reçu au moins un traitement à base de platine.
Objectif(s) secondaire(s)
Evaluer la durée médiane de survie globale dans la meilleure stratégie expérimentale.
Approfondir l’évaluation du profil de sécurité global du meilleur bras expérimental.
Evaluer l’activité anti-tumorale de la meilleure stratégie expérimentale.
Evaluer le bénéfice clinique de la meilleure stratégie expérimentale.
Approfondir l’évaluation de la Survie Sans Progression (PFS) et de la 2e Survie Sans Progression (PFS2 – définie comme le délai entre la randomisation et la seconde progression durant la phase de sélection, et comme le délai entre le début du traitement et la seconde progression durant la phase d’extension) dans la meilleure stratégie expérimentale.
Evaluer les effets des traitements sur les symptômes rapportés par les patientes et la Qualité de vie.
Résumé / schéma de l'étude
Critère(s) d'inclusion
- Carcinosarcome en progression ou récidivant de l’endomètre, (tumeurs de Muller malignes mixtes, MMMT).
- Diagnostic primaire confirmé histologiquement par un anatomopathologiste expert lors d’une revue de la tumeur initiale ou de la biopsie de la maladie en récidive
- Echantillons tumoraux obligatoires : Disponibilité de bloc(s) de tumeur en paraffine du diagnostic de la maladie, ou en cas d’indisponibilité, de la rechute.
- Maladie en progression selon RECIST 1.1.
- Echec après au moins 1 traitement à base de platine, éventuellement administré en traitement adjuvant.
- Patientes ayant reçu un traitement par chimiothérapie pour la prise en charge du carcinosarcome, pouvant inclure une chimiothérapie, une chimiothérapie et une chimio-radiothérapie, et/ou un traitement de consolidation/d’entretien.
- Patientes n’ayant pas d’infection active nécessitant un traitement par antibiotiques.
- Tout traitement hormonal dirigé contre la tumeur maligne doit être interrompu au moins une semaine avant de débuter le traitement du protocole ; la poursuite d’un traitement hormonal de substitution est autorisée.
- Eastern Cooperative Oncology Group (ECOG) ≤ 1.
- Espérance de vie supérieure à 2 mois.
- Fonction hématologique adéquate :
- Nombre de plaquettes ≥ à 100 000/mm3
- Nombre absolu de neutrophiles ≥ 1500/mm3
- Hémoglobine ≥ 9.0 g/dL.
- Fonctions hépatique et rénale adéquates :
- Bilirubine totale ≤ 1.5 x LSN sauf si des métastases hépatiques sont présentes, auquel cas elle doit être ≤ 3 x LSN (≤ 2 chez les patientes atteintes du Syndrome de Gilbert OU bilirubine directe ≤ 1 x LSN).
- Créatinine sérique ≤ 1.5 x LSN ou clairance de la créatinine ≥ 60 mL / min calculée à l’aide de l’équation de Cockcroft-Gault.
- Aspartate aminotransférase (AST) et alanine aminotransférase (ALT) ≤ 2.5 x LSN sauf si des métastases hépatiques sont présentes auquel cas elles doivent être ≤ 5 x LSN
- Phosphatase alcaline ≤ 2.5 x LSN.
- Albumine sérique ≥ 3 g/dL.
- International Normalized Ratio (INR) ou ProThrombin Time (PT) ≤ 1.5 x LSN, et thromboplastine partielle activée (aPTT) ≤ 1.5 x LSN, sauf si les patientes reçoivent un traitement anticoagulant tant que le PT ou la thromboplastine partielle (PTT) sont dans la plage thérapeutique prévue pour l’utilisation d’anticoagulants.
- Pression artérielle normale ou hypertension correctement traitée et contrôlée (pression systolique ≤ 140 mmHg et diastolique ≤ 90 mmHg).
- Les patientes recevant un traitement par corticostéroïdes peuvent poursuivre leur traitement, si leur dose est stable et ≤10mg/jour (dose équivalente prednisone) au minimum depuis 4 semaines avant de débuter le traitement de l’étude.
- Les patientes doivent accepter de ne pas donner leur sang pendant la durée de l’étude et durant les 90 jours suivant la dernière dose de traitement de l’étude.
- Patientes qui ne sont pas en capacité de procréer, ou patientes en capacité de procréer ayant un test de grossesse urinaire ou sérique négatif dans les 72 heures précédant le début du traitement de l’étude, et acceptant de s’abstenir de toute activité pouvant entrainer une grossesse depuis le screening jusqu’à 180 jours après la dernière dose de traitement de l’étude.
- La non-possibilité de procréer est définie comme suit :
- Patientes de 45 ans et plus n’ayant pas eu leurs règles depuis plus d’un an.
- Les patientes ayant une aménorrhée depuis moins de 2 ans sans antécédent d’hystérectomie et d’ovariectomie, doivent avoir un taux d’hormone folliculo-stimulante dans la moyenne post-ménopausique lors de l’examen de screening.
- Post-hystérectomie, ovariectomie post-bilatérale, ou ligature post-tubulaire. L’hystérectomie ou l’ovariectomie doit être confirmée par une procédure documentée dans le dossier médical, ou confirmée par échographie. La ligature des trompes doit être confirmée par une procédure notée dans les dossiers médicaux.
- Pour les patientes en capacité de procréer : les patientes devront accepter d’utiliser une méthode de contraception hautement efficace tout au long de l’étude, depuis le screening jusqu’à 180 jours après la dernière dose de traitement de l’étude. Se rapporter à la section 4.3 du protocole pour la liste des méthodes de contraception hautement efficaces. Les informations doivent être saisies correctement dans les documents sources du centre. Remarque : l’abstinence est acceptable s’il s’agit de la méthode de contraception établie et préférentielle de la patiente.
- La non-possibilité de procréer est définie comme suit :
- Les patientes doivent accepter de ne pas allaiter durant l’étude et pendant 180 jours après la dernière dose de traitement de l’étude.
- Patientes capables de prendre des médicaments par voie orale.
- Patientes de 18 ans ou plus au moment de la signature du consentement.
- Signature du consentement éclairé.
- Pour la France uniquement : Patiente bénéficiaire ou affiliée au système de sécurité sociale
Critère(s) de non-inclusion
- Patientes participant à un autre essai clinique interventionnel (à l’exception d’essais biologiques qui doivent être validés par le promoteur).
- Traitement antérieur par niraparib ou par un autre inhibiteur de PARP ou par des inhibiteurs PD1/PDL-1.
- Patiente ayant reçu un traitement expérimental, une immunothérapie, une chimiothérapie ou une thérapie biologique dans les 4 semaines ou dans un intervalle de temps inférieur à au moins 5 demi-vies du traitement expérimental, selon la durée la plus longue, avant le début du traitement. Patiente ayant eu une radiothérapie dans les 4 semaines avant le début du traitement.
- Chirurgie majeure dans les 3 semaines avant le début du traitement du protocole ou patientes n’ayant pas récupéré complètement des effets secondaires d’une intervention chirurgicale.
- Patientes ayant reçu plus de 3 lignes de traitements systémiques antérieurs pour la prise en charge d’un carcinosarcome utérin.
- Patiente ayant une toxicité persistante, cliniquement significative de grade > 1.
- Maladie cardiovasculaire cliniquement significative (par exemple anomalies significatives de la conduction cardiaque, hypertension non contrôlée, infarctus du myocarde, arythmie cardiaque non contrôlée ou angor instable < 6 mois avant l’inclusion dans l’essai, insuffisance cardiaque congestive de grade ≥ 2 selon la NYHA, arythmie cardiaque grave nécessitant un traitement, pathologie vasculaire périphérique de grade ≥ 2, et antécédent d’accident vasculaire cérébral dans les 6 mois).
- Autre maladie grave qui pourrait accroitre le risque associé à la participation à l’étude ou à l’administration du traitement et qui, selon l’avis de l’investigateur, empêcherait l’inclusion de la patiente dans l’essai, y compris les maladies neurologiques, psychiatriques, infectieuses, hépatiques, rénales, ou gastrointestinales significatives, ou anomalies des examens de laboratoire. Les exemples incluent sans s’y limiter, l’arythmie ventriculaire incontrôlée, infarctus du myocarde récent (dans les 90 jours), crises épileptiques majeures non contrôlées, compression de la moelle épinière instable, syndrome de la veine cave supérieure ou tout trouble psychiatrique qui qui pourrait entraver le recueil du consentement éclairé.
- Symptômes ou signes d’obstruction gastro-intestinale, nécessitant une nutrition ou une hydratation parentérale, ou tout autre trouble ou anomalie gastro-intestinale, y compris une difficulté à avaler, pouvant nuire à l’absorption du médicament.
- Antécédent d’Evènement Indésirable lié au système immunitaire de grade ≥ 3, sous traitement par immunothérapie, à l’exception d’anomalie d’examens de laboratoire cliniquement non-significatives.
- Patiente ayant subi une radiothérapie couvrant plus de 20% de la moelle osseuse dans les 2 semaines précédant le premier jour du traitement du protocole, ou toute radiothérapie dans la semaine précédant le premier jour du traitement du protocole.
- Patiente ayant un diagnostic d’immunodéficience, ou ayant reçu un traitement par stéroïdes systémique >10mg/jour (dose équivalente prednisone) ou toute autre forme de traitement immunosuppresseur dans les 7 jours avant de débuter le traitement de l’essai.
- Les participantes ayant une infection par le VIH connue peuvent être incluses sous réserve des conditions suivantes :
Preuve documentée d’ARN plasmatique du VIH-1 <50 copies/mL de manière persistante, ≤ 3 mois avant le screening de l’étude ET durant le screening. Dans les >3 à 12 mois avant le screening, l’ARN plasmatique du VIH-1 doit être <50 copies/ml de manière constante ; en cas d’augmentations ponctuelles ≥ 50 c/mL, elles ne doivent pas avoir persisté ni être associées à une résistance aux antirétroviraux selon l’investigateur.
ET
Nombre de lymphocytes CD4 >350 cellules/mm3 au cours des 12 derniers mois et lors du screening de l’étude (et aucune mesure ne doit être ≤ 350 cellules/mm3 pendant cette période)
ET
La patiente doit être sous combinaison de traitements thérapeutiques antirétroviraux ininterrompu depuis au moins 3 mois avant le screening, avec un traitement par combinaison d’antirétroviraux conforme aux recommandations.
Les patientes ayant des antécédents d’une maladie de stade 3 CDC (CDC, 2014 ; également appelée syndrome d’immunodéficience acquise) peuvent être incluses si la maladie a été traitée et guérie ou est stable depuis ≥3 mois avant l’entrée dans l’étude ROCSAN. Le sarcome de Kaposi ne nécessitant pas de traitement systémique est autorisé.
Aucun antécédent de lymphome non-hodgkinien associé au VIH ≤ 5 ans avant l’entrée dans l’étude.
Pas de traitement par un vaccin immuno-thérapeutique contre le VIH-1 dans les 90 jours avant le début du screening. - Patiente ayant une hépatite B active (test positif pour l’antigène de surface AgHBs et pour l’anticorps anti-HBc), ou une hépatite C (ARN du VHC détecté) active.
- Maladie auto-immune active qui a nécessité un traitement systémique au cours des 2 dernières années (c’est-à-dire en utilisant des agents modificateurs de la maladie, des corticostéroïdes, ou des immunosuppresseurs). Un traitement de substitution (par exemple la thyroxine, l’insuline, ou les traitements physiologiques de substitution aux corticostéroïdes pour insuffisance surrénale ou hypophysaire, etc) n’est pas considéré comme une forme de traitement systémique.
- Antécédent de pneumopathie interstitielle.
- Administration d’un vaccin vivant dans les 30 jours avant le début du traitement de l’essai.
- Transfusion (plaquettes ou globules rouges) ≤ 4 semaines avant le début du traitement de l’étude.
- Patiente ayant reçu des facteurs colonie-stimulants (par exemple facteur de stimulation des colonies de granulocytes, facteur de stimulation des colonies de granulocytes et de macrophages, ou érythropoïétine recombinante) dans les 4 semaines avant le début du traitement de l’étude.
- Les patientes ne doivent avoir aucun antécédent connu de syndrome myélodysplasique (MDS) ou de leucémie myéloïde aigüe (AML).
- Métastase symptomatique du SNC ou carcinomatose leptoméningée.
- Patiente ayant des antécédents d’autres tumeurs malignes invasives (preuves de la présence d’une autre tumeur maligne au cours des 3 dernières années) ou ayant une tumeur invasive concomitante, à l’exception du cancer de la peau sans mélanome. Les patientes ne sont pas éligibles si leur traitement antérieur contre le cancer contre-indique le traitement de l’étude.
- Réactions d’hypersensibilité connues, ou allergies aux médicaments expérimentaux ou à leurs excipients qui contre-indiquent la participation de la patiente à l’étude.
- Toute considération d’ordre psychologique, familial, sociologique ou géographique pouvant nuire au respect du protocole de l’étude et du calendrier de suivi ; ces considérations devront être discutées avec la patiente avant la signature du consentement de l’étude.
- Patientes faisant l’objet de soins psychiatriques, et les patientes admises dans un établissement sanitaire ou social.
- Patientes privées de liberté par une décision judiciaire ou administrative.
- Patientes faisant l’objet d’une mesure de protection légale ou hors d’état d’exprimer leur consentement.
Calendrier prévisionnel
Lancement de l’étude : Octobre 2024
Fin estimée des inclusions : Octobre 2026
Nombre de patients à inclure : 74
Coordonnateur de l'étude
Pr Isabelle RAY-COQUARD – Centre Léon Bérard – CLCC Lyon
Promoteur de l'étude
ARCAGY-GINECO (Association de Recherche sur les CAncers dont gynécologiques)
