SALVOVAR
Essai pragmatique randomisé de phase III visant à évaluer l’utilité d’un ajustement de la dose et du schéma d’administration de la chimiothérapie chez les patientes dont le cancer de l’ovaire présente des caractéristiques de mauvais pronostic en raison d’une faible chimiosensibilité de la tumeur et d‘une chirurgie d’exérèse ne pouvant être complète
Type d'essai : Académique / Institutionnel
Etat de l'essai : Ouvert
Situation thérapeutique : Tumeur solide ( Adjuvant , Localement avancée / Non résécable , Métastatique / Rechute )
Etablissement(s) participant(s)
Dr Philippe FOLLANA
Dr Rémy LARGILLIER
Dr Julien GRENIER
Dr Nathan AOUIZERAT
Détails de l'essai
Objectif principal
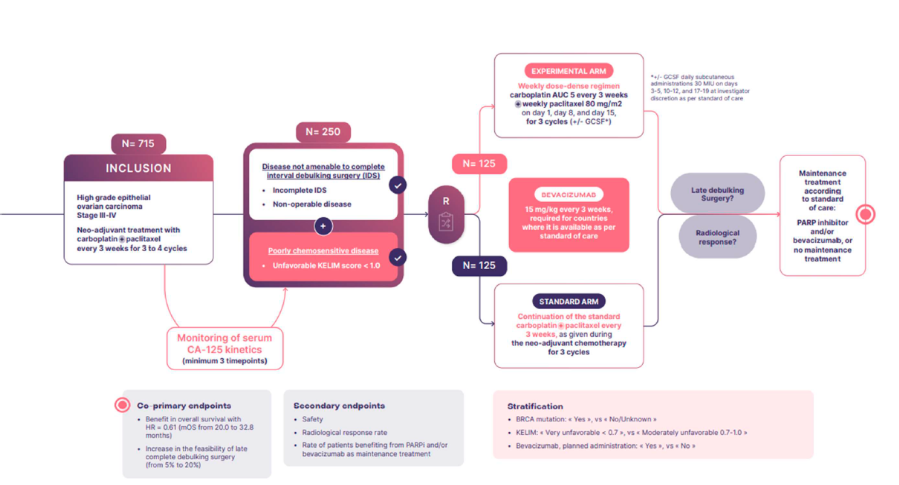
Démontrer une amélioration de l’efficacité de la chimiothérapie de sauvetage avec un schéma dose-dense hebdomadaire du carboplatine paclitaxel par rapport à la poursuite de la chimiothérapie standard par carboplatine-paclitaxel donné toutes les 3 semaines, dans la population
éligible, en termes de :
– Augmentation du pourcentage de chirurgie d’exérèse complète tardive au décours de la chimiothérapie optimisée (augmentation attendue de 5 % à 20 %).
– Augmentation de la survie globale de 49 % (HR = 0,61) dans l’ensemble de la population, se traduisant par une amélioration de la médiane de survie de 20,0 mois (groupe témoin) à 32,8 mois (groupe expérimental) avec une randomisation 1:1.
Objectif(s) secondaire(s)
Comparer l’efficacité de la chimiothérapie de sauvetage dose-dense hebdomadaire par rapport à la poursuite de la chimiothérapie standard avec carboplatine-paclitaxel donné toutes les 3 semaines, en termes de :
• Réponse radiologique après 3 cycles de traitement optimisé, basée sur la meilleure réponse radiologique selon les critères RECIST
• Survie sans progression
Evaluer l’impact de l’adaptation du schéma de chimiothérapie sur le taux de prescription ultérieure d’inhibiteurs de PARP (puisque ces médicaments sont indiqués chez les patientes en réponse complète ou partielle à une chimiothérapie à base de platine) en termes de survie sans progression et de survie globale.
Evaluer l’impact de l’ajout du bevacizumab à la chimiothérapie (bras de sauvetage ou bras standard) sur les résultats en termes d’efficacité sur le taux de réponse radiologique, la survie sans progression, et la survie globale.
Comparer la tolérance avec le bras de chimiothérapie de sauvetage dose-dense hebdomadaire par rapport au bras standard avec carboplatine-paclitaxel toutes les 3 semaines avec/sans bevacizumab, sur les événements indésirables selon le grading NCI-CTCAE.
Résumé / schéma de l'étude
Les patientes seront randomisées selon un ratio 1:1 :
Bras expérimental : Intensification de la dose d’administration de la chimiothérapie et du schéma d’administration du carboplatine-paclitaxel
(carboplatine AUC 5 toutes les 3 semaines et paclitaxel 80 mg/m2 toutes les semaines, au jour 1, jour 8 et jour 15, toutes les 3 semaines) pour 9 doses, potentiellement combinées à des administrations sous-cutanées quotidiennes de GCSF 30 MUI des jours 3 à 5, et 10 à 12, et 17 à 19, à la discrétion de l’investigateur.
Bras standard : Poursuite de la chimiothérapie selon un schéma standard par carboplatine-paclitaxel toutes les 3 semaines, identique à celui administré pendant la chimiothérapie néo-adjuvante.
Critère(s) d'inclusion
- Carcinome épithélial de haut-grade (séreux, endométrioïde ou carcinosarcome avec une composante tumorale épithéliale ≥30%) de l’ovaire, du péritoine primaire ou de la trompe de Fallope, confirmé par l’histologie.
- Patiente âgée de ≥ 18 ans.
- Maladie avancée de stade III ou IV.
- Traitée par 3 à 4 cycles de chimiothérapie néo-adjuvante avec carboplatine-paclitaxel donnés toutes les 3 semaines en première intention, et caractérisée par :
- Un score de KELIMTM défavorable < 1,0 calculé avec l’outil académique KELIM et disponible gratuitement sur le site internet
(https://www.biomarker-kinetics.org/CA-125-neo). - Ne pouvant pas bénéficier d’une chirurgie de cytoréduction d’intervalle complète (tentative de chirurgie de cytoréduction d’intervalle incomplète, ou maladie considérée comme nonopérable).
Il est à noter que l’inclusion de patiente est encouragée en phase de prescreening, c’est-à-dire avant le début de la chimiothérapie néo-adjuvante,
pour permettre d’évaluer prospectivement la cinétique longitudinale du CA125 et l’évaluation de la chirurgie.
- Un score de KELIMTM défavorable < 1,0 calculé avec l’outil académique KELIM et disponible gratuitement sur le site internet
- ECOG PS 0 ou 1.
- Bilan biologique compatible avec une chimiothérapie hebdomadaire dense : globules rouges (baseline hémoglobine ≥ 8 g/dL sans transfusion de globules rouges dans les 3 semaines précédant l’analyse sanguine), globules blancs (numération absolue des neutrophiles ≥ 1500 cellules/mm3) et plaquettes (numération plaquettaire ≥ 100 000/mm3).
- Fonctions rénales et hépatiques adéquates :
- Aspartate aminotransférase (AST) ou alanine aminotransférase (ALT) ≤ 2,5 × LSN, ou ≤ 5 × LSN en cas de métastases hépatiques.
- Bilirubine totale ≤ 1,5 × LSN (les patientes atteints de la maladie de Gilbert sont éligibles si la bilirubine totale ≤ 3 × LSN).
- Albumine ≥3 g/dL.
- Clairance de la créatinine ≥40 ml/min/1,73 m2 (mesurée ou estimée, idéalement avec la formule CKD-EPI sur https://www.kidney.org/professionals/kdoqi/gfr_calculator).
- Les patientes ont donné leur consentement éclairé et écrit pour participer à l’étude.
- Patientes affiliées à un régime d’assurance sociale.
- Patientes désireuses et capables de se conformer au protocole pendant toute la durée de l’étude, y compris le traitement, les visites et examens prévus, y compris le suivi.
Critère(s) de non-inclusion
- Histologie endométrioïde, à cellules claires, mucineuse ou sarcomateuse de bas-grade, ou tumeurs mixtes contenant l’une de ces histologies, ou tumeur ovarienne de bas-grade ou borderline. Pas de contre-indication aux médicaments évalués dans l’essai SALVOVAR (carboplatine, paclitaxel, GCSF).
- Traitement antérieur par bevacizumab au cours de la chimiothérapie néo-adjuvante standard initiale.
- Maladie réfractaire au platine, définie comme une maladie ayant progressé au cours de la chimiothérapie néo-adjuvante.
- Patientes atteintes d’un cancer concomitant, sauf : cancer de la peau sans mélanome traité de manière adéquate, cancer in situ du col de l’utérus traité de manière curative, ou autres tumeurs solides traitées de manière curative sans signe de maladie depuis ≥ 5 ans.
- Traitement par d’autres médicaments expérimentaux dans le cadre d’essais cliniques.
- Affection(s) cliniquement significative(s) non contrôlée(s) qui, de l’avis de l’investigateur, peut/peuvent fausser les résultats de l’essai ou interférer avec la sécurité ou la participation de la patiente, y compris, mais sans s’y limiter, les suivantes :
- Angine de poitrine instable.
- Infarctus du myocarde dans les 6 mois suivant la première dose.
- Maladies concomitantes non contrôlées et/ou graves (hypertension non contrôlée, arythmie de grade ≥ 3 (selon CTCAE v5.0), insuffisance cardiaque, cirrhose).
- Maladie infectieuse active nécessitant un traitement IV (bactéries, virus) dans les 2 semaines précédant la première dose.
- Obstruction de l’œsophage gastrique.
- Obstruction de l’intestin grêle définie par une tomodensitométrie (CT scan) montrant : Boucles dilatées de l’intestin grêle ≤ 12 semaines avant le début de l’étude, ascites/effusions symptomatiques nécessitant une paracentèse ou une thoracentèse ≤30 jours après le début de l’étude.
- Trouble psychiatrique connu qui pourrait interférer avec l’observance de l’essai.
- Patientes enceintes ou allaitantes ou patientes prévoyant de,concevoir des enfants pendant la durée prévue de l’essai.
- Patientes privées de liberté, sous tutelle ou sous curatelle.
Calendrier prévisionnel
Lancement de l’étude : Août 2024
Fin estimée des inclusions : Juin 2027
Nombre de patients à inclure : 250
Coordonnateur de l'étude
Pr Benoit YOU
Hospices civils de Lyon
Promoteur de l'étude
ARCAGY – GINECO