Pan-MSI-AcSé
Dostarlimab en traitement de 1ère ligne chez des patients atteints d’un cancer localement avancé ou métastatique dMMR/MSI (non colorectal/non endométrial) : essai randomisé de phase II avec « Crossover » dans le bras standard à la progression
Type d'essai : Académique / Institutionnel
Etat de l'essai : Ouvert
Situation thérapeutique : Tumeur solide ( Métastatique / Rechute , Localement avancée / Non résécable )
Cibles / marqueurs : MMRd/MSI-H
Etablissement(s) participant(s)
Pr Laetitia DAHAN
Dr Clémence TOULLEC
Détails de l'essai
Objectif principal
L’objectif principal de cet essai est d’évaluer l’efficacité de l’immunothérapie, le dostarlimab, en tant que traitement de première ligne du cancer dMMR/MSI localement avancé ou métastatique non colorectal et non endométrial, par rapport au traitement standard, la chimiothérapie.
Objectif(s) secondaire(s)
Evaluer l’efficacité du traitement dans les deux bras en termes de :
– Taux de réponse objective (TRO),
– Durée de la réponse,
– Survie globale,
– Survie sans progression 2 (SSP2),
– Taux de réponse objective après initiation d’un nouveau traitement anticancéreux (TRO2).
Evaluer la survie sans progression (SSP) chez les patients qui reçoivent le dostarlimab en tant que traitement de deuxième ligne (SSP-c).
Etudier l’association entre l’évolution du taux de l’ADN tumoral circulant (ADNtc) et la survie sans progression dans l’ensemble de la population.
Evaluer la sécurité des traitements.
Evaluer la qualité de vie dans les deux bras à l’aide du questionnaire EORTC QLQ-C30 avant l’initiation de la 2 ème ligne de traitement :
– Proportions de détérioration /de stabilité/d’amélioration à 12 semaines.
– Durée jusqu’à l’apparition d’une détérioration définitive.
Résumé / schéma de l'étude
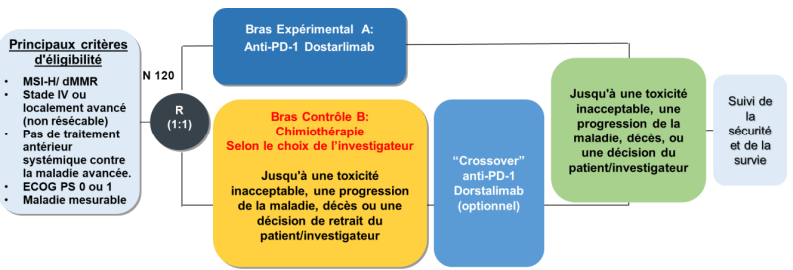
Les patients éligibles seront randomisés en deux groupes :
Groupe expérimental recevant le dostarlimab par voie intraveineuse à une dose de 500 mg toutes les 3 semaines pendant 4 cycles, suivi de 1000 mg toutes les 6 semaines pour tous les cycles suivants.
Groupe contrôle recevant la chimiothérapie conformément aux soins standards.
Le/la patient(e) dont la maladie progresse sous chimiothérapie aura la possibilité de recevoir le traitement expérimental dostarlimab après une période de 28 jours s’il/elle est éligible.
Tous les patients inclus dans l’étude seront traités jusqu’à la progression de la maladie ou jusqu’à leur retrait de l’étude pour quelque raison que ce soit.
Critère(s) d'inclusion
- Le patient devra signer son consentement avant toute autre procédure spécifique à l’essai clinique. Note: en cas d’incapacité du patient à donner son consentement écrit en raison de son état physique, une personne de confiance désignée par le patient, indépendante de l’investigateur ou du promoteur, peut confirmer par écrit le consentement du patient.
- Patients âgés de 18 ans ou plus.
- Maladie localement avancée ou métastatique documentée sans traitement anticancéreux systémique préalable dans ces situations et non éligible à une résection chirurgicale complète.
- Confirmation histologique du statut dMMR/MSI-H des tumeurs solides non colorectales et non endométriales et incluant l’un des cancers suivants : l’adénocarcinome duodénal et de l’intestin grêle, l’adénocarcinome gastrique et de la jonction œso-gastrique avec un score combiné positif (CPS)<5, l’adénocarcinome pancréatique, adénocarcinome ampullaire de Vater, carcinome de la corticosurrénale, carcinome de site primaire inconnu, carcinome neuroendocrine (Grade 3) de toutes origines, et le sarcome des tissus mous à l’exception des tumeurs stromales gastro-intestinales (GIST).
- La thérapie adjuvante pour une maladie non métastatique doit être terminée depuis plus de 6 mois avant le diagnostic de maladie métastatique ou récurrente chez le patient.
- Disponibilité d’au moins 1 bloc de tissu tumoral ou de 20 lames (échantillon d’archivage de moins de 2 ans ou biopsie fraîche de la tumeur primaire et/ou métastatique) pour la confirmation centralisée du statut MMR/MSI par IHC ou NGS/PCR, et pour la recherche translationnelle.
- Les patients présentant une tumeur dMMR/MSI analysée par IHC, PCR (pour l’adénocarcinome gastrique, OGJ et duodénum/intestin grêle uniquement) et/ou NGS dans le centre d’investigation devront être confirmés par une relecture centralisée dans les 24 heures (pour chaque patient, un rapport d’analyse anonymisé sera fourni pour la cette relecture). Les patients ne doivent pas être inclus dans l’étude tant que leur statut dMMR/MSI n’est pas confirmé par le comité de revue.
- Note: En cas de résultat ambigu de l’IHC (absence de contrôle interne positif, perte hétérogène de l’expression des protéines MMR, perte ambiguë d’une seule protéine incluant HMSH6 et PMS2), le statut MSI-H sera évalué par PCR ou NGS pour l’adénocarcinome gastrique, OGJ, duodénum et intestin grêle, et par NGS pour les autres types de tumeurs primaires. Sur la base des résultats de l’IHC et de la PCR ou du NGS (le NGS sera réalisé de manière centralisée dans ce cas), le promoteur décidera de l’inclussion éventuelle des patients.
- Présence d’au moins une lésion mesurable selon les critères RECIST v1.1 dans les 28 jours précédant le début du traitement.
- Statut de performance (ECOG PS) 0-1.
- Statut hématologique: nombre absolu de neutrophiles (NAN) ≥1.5 x 109/L; plaquettes ≥100 x 109/L; hémoglobine ≥ 9 g/dL.
- Note: L’analyse de la formule sanguine complète (FSC) doit être effectuée sans administration de facteurs de stimulation des colonies dans les 4 semaines précédant le prélèvement.
- Fonction rénale adéquate : créatinine sérique <120 μM, ou clairance >50 ml/min (Modification du régime alimentaire dans les maladies rénales [MDRD] ou Cockcroft et Gault).
- Fonction hépatique adéquate : bilirubine sérique ≤ 1,5 x LSN, alanine aminotransférase (ALT) et aspartate aminotransférase (AST) ≤ 3 x LSN, sauf en présence de métastases hépatiques, où elles doivent être ≤ 5 x LSN.
- Pour les patients ne prenant pas de warfarine : INR <1,5 ou TP <1,5 x LSN et soit PTT (temps de thromboplastine) ou aPTT (temps de thromboplastine partielle activée) < 1,5 x LSN. Les participants prenant de la warfarine peuvent être inclus avec une dose stable et un INR thérapeutique < 3,5.
- Les femmes en âge de procréer doivent avoir un test de grossesse sérique négatif dans les 72 heures avant la randomisation.
- Les hommes et les femmes en âge de procréer doivent utiliser une contraception efficace pendant toute la durée de l’étude, ainsi que pendant 4 mois après la dernière dose de dostarlimab (utilisé en première ligne ou en cas de crossover) ou pendant au moins 6 mois après la dernière administration de la chimiothérapie dans le groupe contrôle si aucun passage à dostarlimab n’a eu lieu (selon la dernière version en vigueur du RCP de chaque prduit de chimiothérapie). Les hommes doivent s’abstenir de faire un don de sperme et les femmes doivent s’abstenir de faire un don d’ovocytes pendant cette période spécifiée.
- Patients affiliés à un régime de sécurité social.
- Le patient est disposé et capable de se conformer aux visites prévues, au calendrier de traitement, aux tests de laboratoire, aux biopsies tumorales et à toutes autres exigences de l’étude.
Critère(s) de non-inclusion
- Cancer colorectal et endométrial, ainsi que tout autre cancer primaire non mentionné dans le critère d’inclusion #4.
- Exposition antérieure aux anticorps anti-PD-1, anti-PD-L1 ou anti-CTLA-4 ou traitement par immunothérapie.
- Patients ayant pris un médicament expérimental dans les 4 semaines précédant la première dose dans l’étude (6 semaines pour les anticorps monoclonaux).
- Exposition antérieure à une thérapie anticancéreuse systémique ou à une radiothérapie pour le cancer pour lequel le patient est sélectionné.
- Maladie auto-immune active : Maladie auto-immune active nécessitant un traitement systémique au cours des 2 dernières années (à l’exception d’un traitement de substitution) ou tout antécédent de maladie pulmonaire interstitielle (les patients atteints d’une ancienne maladie auto-immune avec une substitution hormonale orale stable sont éligibles).
- Métastases non contrôlées du système nerveux central, méningite carcinomateuse, autres maladies concomitantes ou infections en cours ou actives.
- Les patients présentant un carcinome gastrique positif pour le récepteur HER2.
- Présence d’une autre maladie grave non maligne non contrôlée ou risque médical élevé dû à un trouble médical grave, une maladie systémique non maligne ou une infection active nécessitant un traitement systémique. Des exemples spécifiques comprennent, mais sans s’y limiter, la présence de pneumonie non infectieuse active, d’arythmie ventriculaire non contrôlée, d’infarctus du myocarde récent (dans les 90 jours), de troubles convulsifs majeurs non contrôlés, de compression instable de la moelle épinière, de syndrome de la veine cave supérieure, ou de tout trouble psychiatrique ou de toxicomanie qui pourrait entraver la coopération avec les exigences de l’étude.
- Transplantation antérieure de moelle osseuse allogénique ou d’organe solide.
- Les patients ayant reçu un traitement avec des corticostéroïdes systémiques ou d’autres médicaments immunosuppresseurs systémiques (incluant, sans s’y limiter, la prednisone, la dexaméthasone, le cyclophosphamide, l’azathioprine, le méthotrexate, la thalidomide et les agents anti-facteur de nécrose tumorale) dans les 2 semaines précédant la première dose de traitement adjuvant ou nécessitant un traitement immunosuppresseur systémique pendant l’étude. Les stéroïdes inhalés ou topiques ainsi que les doses de substitution par glucocorticoïde >10 mg d’équivalent prednisone par jour sont autorisés en l’absence de maladie auto-immune active.
- Note 1: Les patients ayant reçu des médicaments immunosuppresseurs systémiques à dose faible (par exemple, une dose unique de dexaméthasone pour les nausées) peuvent être inclus dans l’étude après l’approbation du médecin.
- Note 2: Utilisation autorisée de corticostéroïdes topiques, oculaires, intra-articulaires, intranasaux et inhalés avec absorption systémique minimale. Des doses de substitution par glucocorticoïde, y compris >10 mg par jour de prednisone sont autorisées. Une cure brève (moins de 3 semaines) de corticostéroïdes pour prophylaxie (par exemple allergie aux produits de contraste) ou traitement de maladies non auto-immunes (par exemple réaction d’hypersensibilité retardée causée par un allergène de contact) est autorisée.
- Toute autre malignité concomitante ou antérieure différente de la maladie de l’étude, à l’exception des cas suivants : i. Carcinome in situ du col de l’utérus traité de manière adéquate, ii. Carcinome basocellulaire ou épidermoïde de la peau, iii. Cancer en rémission complète depuis plus de 2 ans.
- Infection connue par le virus de l’immunodéficience humaine (VIH).
- Le patient ayant reçu un vaccin vivant dans les 14 jours précédents.
- Le patient avec une présence documentée de l’antigène de surface du virus de l’hépatite B HBsAg [ou l’anticorps du virus de l’hépatite B HBcAb] lors de la visite de pré-inclusion ou dans les 3 mois précédant l’administration initiale du traitement de l’étude.
- Le participant positif à l’anticorps du virus de l’hépatite C anti-VHC lors de la visite de pré- inclusion ou dans les 3 mois précédant la première dose du traitement de l’étude.
- Note: les participants positifs à l’anticorps anti-VHC en raison d’une maladie résolue peuvent être inclus, à condition d’obtenir un test négatif confirmatoire de l’ARN du VHC. Le participant positif au test de l’ARN du VHC lors de la visite de pré-inclusion ou dans les 3 mois précédant la première dose de traitement de l’étude. Note : Le test de l’ARN du VHC est facultatif et les participants ayant un résultat négatif à l’anticorps anti-VHC ne sont pas obligés de passer également le test de l’ARN du VHC.
- Hypersensibilité antérieure sévère connue au produit expérimental ou à l’un de ses excipients.
- Femmes enceintes ou allaitantes.
- Participation à une autre étude clinique dans les 30 jours précédant la première administration du traitement de l’étude ou pendant l’étude.
- Présence de toute condition psychologique, familiale, sociologique ou géographique entravant potentiellement le respect du protocole de l’étude et du calendrier de suivi.
- Personne privée de sa liberté ou placée sous protection légale ou sous tutelle. Les patients assignés au groupe de traitement standard (Bras B) peuvent recevoir le dostarlimab (Bras A) en cas de progression documentée de la maladie selon les critères RECIST v1.1 (patients en « crossover »).
Les critères d’inclusion et d’exclusion pour le passage sont identiques, à l’exception des critères d’inclusion #3 et #4.
Critère #3 pour le crossover est : Patient inclus et randomisé dans le groupe “contrôle” avec une maladie progressive documentée selon les critères RECIST v1.1.
Le critère #4 pour le crossover est : Exposition antérieure à une chimiothérapie pour une maladie localement avancée ou métastatique.
Calendrier prévisionnel
Lancement de l’étude : Mai 2024
Fin estimée des inclusions : Novembre 2026
Nombre de patients à inclure : 120
Coordonnateur de l'étude
Pr Thierry ANDRE – Hôpital Saint Antoine -AP-HP
Promoteur de l'étude
UNICANCER