Prodige 95 - CIME
Etude multicentrique, randomisée, comparative, menée en ouvert, de phase III visant à comparer la survie des patients atteints d’adénocarcinomes œsogastriques localement avancés ou métastatiques, MSI/dMMR, traités par une combinaison d’immunothérapie (botensilimab + balstilimab) versus le traitement standard (FOLFOX/XELOX + nivolumab)
Phase : III
Type d'essai : Académique / Institutionnel
Etat de l'essai : Ouvert
Situation thérapeutique : Tumeur solide ( Métastatique / Rechute , Localement avancée / Non résécable )
Cibles / marqueurs : PDL1 / MMRd/MSI-H
Type d'essai : Académique / Institutionnel
Etat de l'essai : Ouvert
Situation thérapeutique : Tumeur solide ( Métastatique / Rechute , Localement avancée / Non résécable )
Cibles / marqueurs : PDL1 / MMRd/MSI-H
Etablissement(s) participant(s)
Dr Christelle DE LA FOUCHARDIERE
Détails de l'essai
Objectif principal
Survie globale (SG).
Objectif(s) secondaire(s)
Survie sans progression (SSP), taux deréponse objective après 16 semaines de traitement (TRO-16S) et durée de réponse selon RECIST V1.1.
Evénements indésirables (EI), EIs reliés aux traitements, EI liés aux traitements et entraînant une réduction de la dose ou un arrêt du traitement, événements indésirables graves (EIG), événements indésirables grave inattendus(EIGI), et EI immuno- induits gradés selon NCICTCAE V5.0
Evolution de la qualité de vie des patients selon le questionnaire EORTC QLQC30.
Résumé / schéma de l'étude
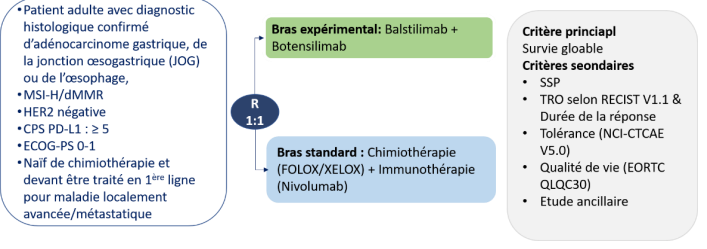
Critère(s) d'inclusion
- Homme ou femme âgé(e) ≥ 18 ans à la date de signature du consentement.
- Patients avec diagnostic histologique confirmé d’adénocarcinome gastrique, de la jonction œsogastrique (JOG) ou de l’œsophage, HER2 negative, présentant une instabilité des microsatellites/déficience de la réparation des mésappariements (MSI/dMMR) dont les tumeurs expriment PD-L1 avec un score positif combiné (CPS) ≥ 5.
- Patient devant être traité avec une thérapie de première ligne en phase localement avancée ou métastatique.
- Patient sans aucun traitement antérieur par chimiothérapie en phase localement avancée ou métastatique.
- Patient avec maladie mesurable (en dehors de toute zone irradiée dans les 6 derniers mois), définie comme au moins une lésion unidimensionnelle qui peut être mesurée avec précision ≥ 10 mm avec une scanner (CT scan) selon RECIST V1.1.
- Patient ayant un indice de performance ECOG de 0 ou 1.
- Patient avec fonctions hématologiques et des principaux organes adéquats tels que :
- Numération absolue des neutrophiles ≥ 1,5 109/L (sans apport de facteurs de croissance dans les 14 jours)
- Plaquettes ≥ 100 109/L (sans transfusion de plaquettes dans les 7 jours)
- Hémoglobine ≥ 9 g/dL (sans transfusion dans les 7 jours)
- Clairance de la créatinine selon CKD-EPI ≥ 30 mL/min/1,73 m2
- Bilirubine totale sérique ≤ 1,5 x LSN (sauf pour les patients atteints de la maladie de Gilbert pour lesquels une bilirubine totale sérique ≤ 3 x LSN est acceptable)
- ASAT et ALAT ≤ 3 x LSN (ou jusqu’à 5 x LSN en cas de métastase hépatique ou d’infiltration hépatique)
- Disponibilité d’un échantillon représentatif de tissu tumoral de la tumeur primitive ou de métastase fixé au formol et inclus en paraffine (FFPE) (pièce opératoire ou biopsie) avec le compte rendu pathologique correspondant. Cet échantillon tumoral doit répondre aux critères de contrôle qualité/quantité suivants : ≥ 30% de cellules tumorales et une surface tumorale ≥ 5mm² (ou une maladie biopsiable selon le critère d’inclusion suivant).
- Patient avec au moins 1 lésion tumorale visible par imagerie médicale et accessible à un prélèvement percutané ou endoscopique de façon répétée permettant la réalisation d’une biopsie sans risque inacceptable de complications et appropriée pour le prélèvement d’un minimum de 4 cores avec une aiguille d’un diamètre minimum de 16 gauge.
- Les femmes en âge de procréer doivent justifier d’un test de grossesse sérique négatif lors de la visite de screening (dans les 72 heures avant la première dose
des traitements de l’étude) et doivent accepter d’utiliser des mesures contraceptives hautement efficaces à compter du jour de la visite de screening jusqu’à.
• 9 mois après la fin du traitement par oxaliplatine
• 6 mois après la fin du traitement par fluorouracile
• 5 mois après la fin du traitement par nivolumab ou botensilimab ou Balstilimab
• 6 mois après la fin du traitement par capecitabine - Les hommes ayant une partenaire féminine en âge de procréer doivent accepter d’utiliser des mesures contraceptives hautement efficaces pendant toute la durée de l’étude, à partir de la visite de screening jusqu’à 6 mois après la fin du traitement par oxaliplatine ou 3 mois après la dernière dose des autres traitements de l’étude. Les hommes ayant des partenaires enceintes doivent accepter d’utiliser un préservatif ; aucune mesure contraceptive supplémentaire n’est requise pour la partenaire enceinte.
- Le patient doit comprendre, signer et dater le consentement éclairé avant toute procédure spécifique au protocole et doit être capable et disposé à se conformer aux visites et procédures de l’étude imposées par le protocole.
- Patient affilié ou bénéficiaire d’un régime de sécurité sociale.
Critère(s) de non-inclusion
- Patient avec un cancer œsogastrique éligible à un traitement curatif.
- Patient précédemment traités par anti-PD-1, anti-PD-L1 ou anti-CTLA-4 ou toute autre immunothérapie.
- Patients avec chirurgie ou radiothérapie dans les 4 semaines précédant le C1J1
- Patients avec des EI de Grade >1 persistant et relies aux précédents traitements anti-cancer, à l’exception de l’alopécie (tot grade autorisé) et des valeurs biologiques présentées en critère I7.
- Patients avec kaliémie, magnesémie et calcémie inférieures à la norale ,
- Patients avec une prolongation connue de l’intervalle QT/QTc : intervalle QT/QTc supérieur à 450 msec pour les hommes et supérieur à 470 msec pour les femmes selon l’inclusion de l’ECG.
- Patient avec métastases du système nerveux central (SNC) symptomatiques, non traitées ou en progression active.
- Patient atteints d’un autre cancer, sauf si ce cancer n’est pas susceptible d’interférer avec les résultats de cette étude (eg :carcinome basocellulaire ou squameux de la peau, carcinome in situ du col de l’utérus, cancer localisé de la prostate) ou en l’absence de maladie depuis ≥ 2 ans.
- Patient avec allergie ou hypersensibilité connue à l’un des médicaments de l’étude ou à l’un des excipients des médicaments de l’étude.
- Patient avec documentation de maladie pulmonaire interstitielle (MPI) actuelle ou de pneumonite, ou antécédents de MPI ou de pneumonite non infectieuse nécessitant des glucocorticoïdes.
- Patient avec antécédents de transplantation d’organe allogénique.
- Patient ave troubles psychiatriques ou liés à la consommation de stupéfiants
- Patient avec neuropathie périphérique avec impact fonctionnel
- Patients avec pathologie cardiaque active ou infarctus du myocarde dans les 6 mois, ou antécédent de pathologie cardiaque non contrôlée.
- Patient avec un traitement récent (dans les 7 jours précèdent C1J1) ou concomitant à venir avec la brivudine.
- Patient avec déficit compet de l’activité de la dihydropyrimidine déshydrogénase (DPD) avec valeur uracile sanguin ≥ 150 ng/mL) ou partiel : valeur uracile sanguin ≥ 16 ng/ml et < 150 ng/mL. qui pourraient altérer la coopération du patient avec les exigences de l’étude.
- Patient atteint d’une affection nécessitant un traitement systémique par des corticostéroïdes (> 10 mg/jour d’équivalent prednisone) dans les 14 jours avant la première dose des traitements de l’étude ou un autre médicament immunosuppresseur dans les 30 jours avant la première dose des traitements de l’étude. Les stéroïdes inhalés ou topiques et les doses de stéroïdes (≤ 10 mg/jour d’équivalent prednisone) sont autorisés en l’absence de maladie auto-immune active.
- Patient ayant reçu un vaccin vivant dans les 4 semaines avant C1J1.
- Patient avec maladie auto-immune active ou antécédents de maladie autoimmune ayant nécessité un traitement systémique dans les 2 ans avant le début du traitement de l’étude (c’est-à-dire avec l’utilisation d’agents modificateurs de maladie ou de médicaments immunosuppresseurs).
- Patient avec antécédents ou preuve actuelle de toute affection, condition, comorbidité, thérapie, infection active ou anomalie biologique pouvant fausser les résultats de l’étude, interférer avec la participation du patient pour la durée complète de l’étude ou s’il n’est pas de ce fait dans le meilleur intérêt du patient de participer, selon l’avis de l’investigateur.
- Patients présentant une infection documentée:
• hépatite B active (chronique ou aiguë ; définie par un test positif d’antigène de surface de l’hépatite B [HBsAg] lors du screening), sauf si leur VHB est contrôlé de manière stable par des analogues nucléosidiques (comme l’entécavir ou le ténofovir) qui seront poursuivis pendant toute la durée de l’étude.
• hépatite C active. Les patients positifs pour l’anticorps de virus de l’hépatite C (VHC) ne sont éligibles que si le PCR est négatif pour l’ARN du VHC.
• VIH - Patient avec transplantation antérieure d’organe ou de moelle osseuse.
- Femmes enceintes ou allaitantes.
Calendrier prévisionnel
Lancement de l’étude : Décembre 2025
Fin estimée des inclusions : Décembre 2027
Nombre de patients à inclure : 132
Coordonnateur de l'étude
Dr Clélia COUTZAC – Centre Léon Bérard – CLCC Lyon
Promoteur de l'étude
Centre Léon Bérard – CLCC Lyon
Dernière mise à jour le 22 avril 2026