GCO-003 TARLANEC
Etude randomisée de phase III comparant le tarlatamab à une chimiothérapie standard chez des patients pré-traités présentant un carcinome neuroendocrine pulmonaire ou gastro-entéro-pancréatique peu différencié de stade avancé
Type d'essai : Académique / Institutionnel
Etat de l'essai : Ouvert
Situation thérapeutique : Tumeur solide
Etablissement(s) participant(s)
Pr Laurent GREILLIER
Pr Patricia NICCOLI
Pr Olivier BYLICKI
Dr Nicolas CLOAREC
Détails de l'essai
Objectif principal
Evaluer l’efficacité du tarlatamab chez les patients présentant un carcinome neuroendocrine pulmonaire ou gastro-entéro-pancréatique peu différencié de stade avancé après traitement par
une chimiothérapie à base de platine mesurée par la survie globale (OS) après au moins une dose de traitement.
Objectif(s) secondaire(s)
Evaluer la sécurité du traitement : incidence, nature et gravité des évènements indésirables (CTCAE v5.0 ; ASTCT 2019 pour le syndrome de relargage des cytokines (CRS) et le syndrome de neurotoxicité associé aux cellules immunitaires effectrices (ICANS)).
Evaluer l’efficacité du traitement mesurée par :
o Le taux de réponse objective selon RECIST 1.1 (ORR),
o La durée de réponse (DoR),
o Le taux de contrôle de la maladie (DCR),
o La survie sans récidive (PFS),
o La survie globale (OS).
Evaluer la qualité de vie des patients.
Résumé / schéma de l'étude
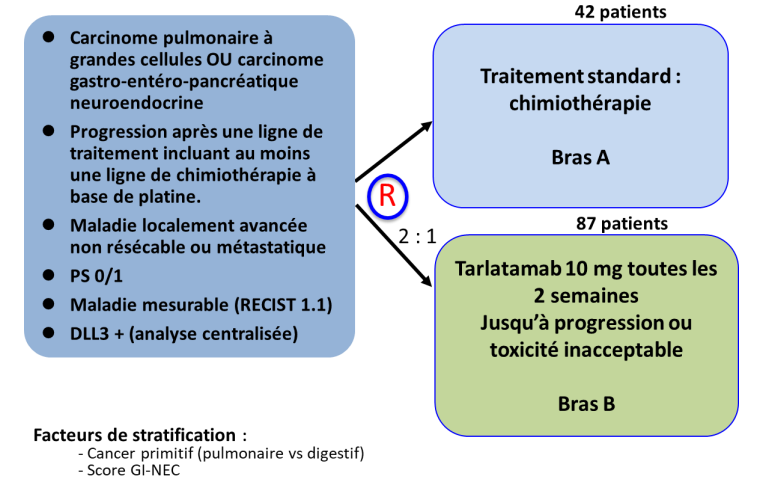
Critère(s) d'inclusion
- Consentement éclairé, écrit et signé :
- Les patients doivent avoir signé et daté le formulaire de consentement éclairé écrit approuvé par le comité d’éthique en accord avec le cadre juridique et institutionnel. Il doit avoir été signé avant que des procédures liées au protocole ne faisant pas partie de la prise en charge standard des patients soient réalisées.
- Les patients doivent être disposés et capables de respecter le calendrier des visites, du traitement et des analyses de laboratoire.
- Age ≥ 18 ans.
- WHO Performance Status PS 0-1.
- Espérance de vie > 12 semaines.
- Carcinome neuroendocrine (CNE) peu différencié prouvé histologiquement avec confirmation par le laboratoire centralisé :
- CNE pulmonaire à grandes cellules (classification OMS 2015),
ou - CNE gastro-entéro-pancréatique à petites et grandes cellules (évalué sur des tissus archivés, possibilité de pré-screening durant la 1ère ligne de traitement).
- CNE pulmonaire à grandes cellules (classification OMS 2015),
- Expression de DLL3 dans au moins 1% des cellules tumorales (évaluée sur des tissus archivés, possibilité de pré-screening durant la 1ère ligne de traitement).
- Progression tumorale après une ligne de chimiothérapie à base de platine.
- Maladie localement avancée non résécable ou métastatique.
- Au moins une lésion cible mesurable selon RECIST 1.1 selon l’évaluation de l’investigateur. L’évaluation radiologique doit être faite dans les délais indiqués dans le protocole.
- Fonction biologique adéquate :
- Clairance de la créatinine > 50 mL/min
- Neutrophiles ≥ 1 500/mm3
- Plaquettes > 100 000/mm3
- Hémoglobine > 9 g/dL
- AST et ALT < 3 x LNS avec bilirubine totale ≤ 2 × LNS, à l’exception des patients présentant un syndrome de Gilbert documenté ou des métastases hépatiques, qui doivent avoir des AST et ALT ≤ 5 x LNS et une bilirubine totale ≤ 3,0 mg/dL.
- Les patients doivent avoir récupéré de toutes les toxicités associées au traitement antérieur jusqu’à un état de base acceptable ou un grade 0-1 selon le NCI CTCAE v5.0, à l’exception
des toxicités qui ne sont pas considérées comme un risque pour la tolérance comme l’alopécie ou le vitiligo. - Matériel tumoral disponible pour l’analyse centralisée et les projets de recherches translationnelles.
- Absence de maladie non contrôlée et facteurs psychologiques, familiaux, sociologiques ou géographiques susceptible de compromettre le respect du protocole et le calendrier de suivi
de l’étude. - Les femmes en âge de procréer qui sont sexuellement actives avec un partenaire masculin non stérilisé doivent utiliser une méthode de contraception hautement efficace pendant les
28 jours précédant la première administration du traitement à l’étude et jusqu’à 7 mois après la dernière dose du traitement à l’étude. L’arrêt de la contraception après ce délai doit être discuté avec un médecin. L’abstinence périodique, la méthode du rythme et la méthode du retrait de ne sont pas des méthodes de contraception acceptables. Les femmes doivent s’abstenir d’effectuer des dons d’ovocytes pendant les 7 mois après la dernière dose du traitement à l’étude. - Les hommes qui sont sexuellement actifs avec des femmes en âge de procréer devront respecter une méthode de contraception pendant une période de 6 mois après la dernière
dose de traitement. - Le patient doit être couvert par un régime national d’assurance maladie.
Critère(s) de non-inclusion
- Tumeur neuroendocrine bien différenciée (TNE G1, G2 et G3 selon la classification digestive OMS 2027 ou tumeur carcinoïde typique/atypique selon la classification pulmonaire OMS 2015).
- Traitement antérieur ciblant DLL3.
- Plus d’une ligne de traitement systémique au stade métastatique. La chimiothérapie au stade non métastatique n’est pas considérée comme une 1ère ligne s’il y a au moins un intervalle de 6 mois entre la dernière dose de chimiothérapie au stade non métastatique et l’instauration d’une chimiothérapie de 1ère ligne pour la maladie métastatique/rechute.
- CNE pulmonaire à petites cellules (sauf en tant que composante mineure < 30% dans les tumeurs mixtes).
- Connaissance d’une mutation activatrice de l’EGFR ou réarrangement ALK ou ROS1 en cas de CNE pulmonaire.
- Métastases du système nerveux central (SNC) non traitées ou symptomatiques. Métastases asymptomatiques du SNC : les patients sont éligibles s’ils sont cliniquement stables depuis au moins 4 semaines et ne nécessitent pas d’intervention (y compris l’utilisation de corticostéroïdes).
Métastases traitées du SNC :
– Les patients sont éligibles si les métastases cérébrales sont asymptomatiques.
– La radiothérapie cérébrale in toto ou la chirurgie est terminée depuis au moins 2 semaines avant la première dose du traitement à l’étude (la radiochirurgie stéréotaxique a été effectuée au moins 7 jours avant la première dose du traitement à l’étude).
Les métastases du CNS sont cliniquement stables si :
– Le patient n’a pas pris de corticoïdes pour les métastases du CNS depuis au moins 5 jours (sauf si les corticoïdes sont prescrits pour une raison autre que les métastases du CNS).
– Le patient n’a pas pris de médicaments antiépileptiques ou a pris des doses stables depuis au moins 14 jours avant la première dose de traitement à l’étude. - Métastases leptoméningées.
- Patients ayant des antécédents récents d’autres tumeurs malignes, à l’exception du cancer de la peau non mélanome traité de manière adéquate et du cancer in situ traité de manière
curative. Les patients ayant des antécédents de tumeurs solides, y compris d’adénocarcinome, traité de manière curative avec ou sans chimiothérapie et sans aucun signe de la malade depuis > 2 ans avant la randomisation peuvent être inclus. - Chirurgie majeure dans les 28 jours avant l’initiation du traitement à l’étude.
- Infarctus du myocarde et/ou insuffisance cardiaque congestive symptomatique (classe > II New York Head Association) dans les 12 mois précédant le début du traitement à l’étude.
- Antécédents de thrombose artérielle (par exemple, accident vasculaire cérébral ou accident ischémique transitoire) dans les 12 mois précédant le début du traitement à l’étude.
- Symptômes et/ou signes cliniques et/ou radiologiques suggérant une infection systémique active non contrôlée et/ou aiguë dans les 7 jours précédant la première administration du
traitement à l’étude. Si un patient présente une infection active nécessitant une antibiothérapie parentérale, dès que cette antibiothérapie est terminée et que les symptômes
ont disparu, le patient peut être considéré comme éligible. - Sensibilité connue et/ou hypersensibilité immédiate à l’un des composants du traitement à l’étude.
- Antécédents d’immunodéficience primaire, antécédents de transplantation d’organe nécessitant une immunosuppression thérapeutique et l’utilisation d’agents immunosuppresseurs dans les 28 jours précédant la randomisation ou antécédents de toxicité sévère (grade 3 ou 4) à médiation immunitaire résultant d’une autre thérapie
immunitaire. - Patients ayant présenté une pneumopathie inflammatoire, des troubles de l’hypophyse ou de la thyroïde, ou une pancréatite sous traitement par des agents immuno-oncologique.
- Patients signalant des réactions liées à la perfusion ou des évènements indésirables grave à médiation immunitaire, mettant en jeu le pronostic vital ou récurrents (grade ≥ 2), y compris des évènements entraînant l’arrêt définitif des agents immuno-oncologique.
- Présence d’une sonde à demeure ou d’un drain (y compris les éléments suivants : tube de néphrostomie percutanée, sonde de Foley à demeure, drain biliaire, drain ou cathéter
péritonéal, drain ou cathéter péricardique, cathéter de drainage ou drain thoracique pour la collecte de liquide pleural). - Patient présentant un diagnostic d’immunodéficience ou en cours de traitement par une corticothérapie systémique ou toute autre forme de traitement immunosuppresseur dans les
7 jours précédant l’administration de la première dose du traitement à l’étude. - Hépatite B ou C aiguë connue par évaluation sérologique. Les patients présentant des séquelles sérologiques d’hépatite (test d’anticorps sérologique positif pour le virus) sans
hépatite peuvent être inclus. - Infection connue par le virus de l’immunodéficience humaine (VIH).
- Patiente enceinte ou allaitante, ou qui prévoit de l’être durant l’étude et dans les 7 mois après la dernière administration du traitement à l’étude.
- Homme ne souhaitant pas s’abstenir de faire un don de sperme durant l’étude et dans les 6 mois après la dernière administration du traitement à l’étude.
- Vaccination par des virus vivants ou atténués non autorisée pendant les 28 jours précédant l’administration de la 1ère dose du traitement à l’étude et pendant toute la durée de l’étude. La vaccination contre le coronavirus 2 du syndrome respiratoire aigu sévère (SARS-CoV-2) doit être évitée durant le screening et au moins 14 jours avant le 1er jour de traitement. Les vaccins antivarioliques vivants et non réplicatifs (tels que Jynneos) contre la variole du singe sont autorisés pendant l’étude (sauf pendant le 1 er cycle) conformément aux pratiques habituelles du centre et aux recommandations internes.
- Maladie auto-immune active nécessitant un traitement systémique (à l’exception d’un traitement de substitution) au cours des deux dernières années ou toute autre maladie
nécessitant un traitement immunosuppresseur pendant l’étude. - Patients souffrant d’une autre maladie concomitante grave et/ou non contrôlée qui pourrait compromettre la participation à l’étude
Calendrier prévisionnel
Lancement de l’étude : Septembre 2025
Fin estimée des inclusions : Mars 2028
Nombre de patients à inclure : 129
Information(s) complémentaire(s)
BIO-IFCT2404
Prélèvements tissulaires
Un échantillon tissulaire à l’inclusion pour l’analyse centralisée de DLL3 :
– Bloc FFPE permettant d’effectuer au moins 6 lames de 4 µm (recommandé) et lames HES correspondante.
– 6 lames blanches de 4 µm chargées positivement et lames HES correspondante. Les lames doivent être fraichement coupées.
Prélèvements sanguins
Des échantillons sanguins seront prélevés à l’inclusion, à 8 semaines et à progression. 3 tubes EDTA de 6 mL et 1 tube sec CAT 6 mL seront prélevés à chaque point de prélèvement.
Coordonnateur de l'étude
Pr Nicolas GIRARD – Institut Curie
Promoteur de l'étude
Intergroupe Francophone de Cancérologie Thoracique (IFCT)