DEFINITIVE
Traitement diagnostique guidé par HER2DX chez les patients présentant un cancer du sein HER2-positif de stade précoce
Type d'essai :
Etat de l'essai : Ouvert
Situation thérapeutique : Tumeur solide ( Néoadjuvant )
Cibles / marqueurs : HER2+
Etablissement(s) participant(s)
Dr Alexandre TASSIN DE NONNEVILLE
Dr Julien GRENIER
Détails de l'essai
Objectif principal
Déterminer une éventuelle amélioration de la qualité de vie liée à la santé (QVLS) suite à un traitement guidé par le test HER2DX, comparé au traitement de référence.
Evaluer si la stratégie d’adaptation du traitement guidé par HER2DX présente un taux de réponses similaire à celui du traitement standard.
Objectif(s) secondaire(s)
Evaluer d’autres critères de jugement rapportés par les patients (valeurs fonctionnelles et symptomatiques) après un traitement personnalisé par HER2DX par rapport au traitement standard.
Evaluer si la stratégie d’adaptation du traitement guidé par HER2DX présente des résultats d’efficacité similaires à ceux du traitement standard.
Evaluer l’association entre les scores HER2DX et les résultats d’efficacité.
Evaluer l’innocuité et la tolérance du traitement guidé par test et du traitement standard correspondant.
Evaluer l’expérience du patient dans le bras témoin par rapport à ceux traités à l’aide du test et évaluer si l’inclusion du test HER2DX a un impact sur l’expérience du patient.
Analysez l’impact financier du test HER2DX.
Evaluer si la désescalade potentielle du traitement après un traitement adapté par HER2DX pourrait avoir un impact sur la productivité du travail.
Résumé / schéma de l'étude
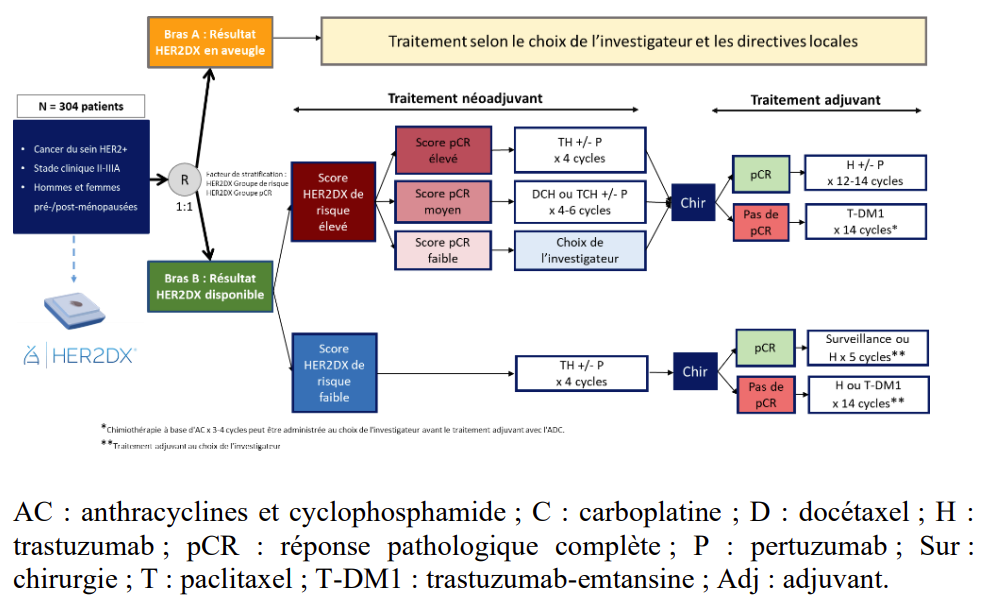
Critère(s) d'inclusion
- Un consentement éclairé signé doit être obtenu avant toute procédure spécifique à l’essai.
Remarque : Les patients candidats en France doivent être affiliés à un système de Sécurité sociale (ou équivalent). - Patients hommes/femmes âgés d’au moins 18 ans le jour de la signature du consentement éclairé.
- Avoir un indice de performance ECOG (Eastern Cooperative Oncology Group) de 0 à 2.
- Eligible à un traitement par taxane, carboplatine, trastuzumab, pertuzumab et T-DM1.
- Adénocarcinome invasif primitif du sein, non métastatique, confirmé histologiquement, non traité et récemment diagnostiqué.
- Stade à la consultation : cT1 cN1-2 ou cT2-3 cN0-2 tel que déterminé par le système de stadification AJCC, 8e édition (spécifiquement conformément aux règles du groupe de stade anatomique).
Remarque : Le statut des ganglions lymphatiques axillaires doit être évalué par biopsie à l’aiguille fine ou biopsie au trocart. Cette procédure à la sélection sera omise en cas d’absence de suspicion radiographique de ganglion(s) axillaire(s) positif(s) ou si un compte-rendu des résultats d’une biopsie à l’aiguille fine ou d’une biopsie au trocart concernant des ganglions lymphatiques suspects est disponible avant la période de sélection. - Absence de métastases à distance (c.-à-d. cM0).
- Les patients atteints de tumeurs multifocales (plus d’une masse confinée dans le même quadrant que la tumeur primaire) sont éligibles à condition qu’au moins un foyer soit échantillonné et localement confirmé comme HER2- positif.
- Les patients atteints de tumeurs multicentriques (tumeurs multiples impliquant plus d’un quadrant) sont éligibles à condition que toutes les lésions isolées soient échantillonnées et confirmées localement comme HER2- positives.
Remarque : Chez les patients atteints d’un cancer du sein multifocal ou multicentrique, la lésion la plus importante doit être mesurée pour déterminer le stade T et pour effectuer le test HER2DX. - La positivité HER2 est définie comme suit : IHC 3+ ou HER2 2+/ ISH positif conformément aux dernières directives ASCO/CAP selon le laboratoire local, sur la base de l’échantillon de tissu analysé le plus récemment.
- Le statut ER/PR est déterminé localement sur la base du matériel de biopsie mammaire de prétraitement conformément aux directives ASCO/CAP les plus récentes.
- Éligibles à un traitement néoadjuvant.
- Le patient accepte de subir une prise en charge chirurgicale appropriée, y compris une chirurgie des ganglions lymphatiques axillaires et une mastectomie partielle ou totale, après la fin du traitement néoadjuvant.
- La fraction d’éjection ventriculaire gauche (FEVG) à la baseline ≥ 50 %, mesurée par échocardiogramme (ECHO) ou par acquisition multiple (MUGA).
- Disponibilité (avant traitement) d’un échantillon de tissu tumoral provenant d’un bloc tumoral FFPE de la tumeur primaire du sein pour le test diagnostique HER2DX. Le tissu tumoral doit être de bonne qualité sur la base du contenu tumoral total et viable et sa qualité doit être évalué de manière centralisée avant l’inclusion.
Remarque : Un échantillon FFPE archivé représentatif de la tumeur primitive dans le sein, ainsi qu’un rapport de pathologie documentant la positivité de HER2 et le statut ER/PR, seront envoyés au laboratoire central. S’il n’est pas disponible ou s’il est insuffisant en termes de quantité ou de qualité, d’autres matériels tumoraux peuvent être envoyés au laboratoire central. - Fonction hématologique et des organes cibles adéquats :
- Nombre absolu de neutrophiles (ANC) ≥ 1500/mm3 ou ≥1,5 × 109 /L (le soutien du facteur de stimulation des colonies est autorisé).
- Numération plaquettaire ≥ 100 000/μL (les transfusions de plaquettes ne sont pas autorisées dans les 14 jours précédant le cycle 1 jour 1 pour répondre aux critères d’admissibilité).
- Hémoglobine (Hgb) ≥ 9,0 g/dL ou ≥ 6,21 mmol/L (le soutien du facteur de stimulation des colonies est autorisé).
- Créatinine OU mesurée ou clairance de la créatinine (calculée à l’aide de la formule de Cockcroft-Gault) ≤ 1,5 × LSN OU ≥ 30 mL/min pour les patients présentant des taux de créatinine >1,5 × LSN en établissement.
- Bilirubine totale Bilirubine totale ≤ 1,5 × LSN, sauf pour les patients atteints du syndrome de Gilbert qui ne peuvent être inclus que si la bilirubine totale est ≤ 3,0 × LSN ou la bilirubine directe ≤ 1,5 × LSN.
- AST (SGOT) et ALT (SGPT) ≤ 3 × LSN.
- Rapport international normalisé (INR) ou temps de prothrombine (TP) Temps de thromboplastine partielle activée (TCA) ≤1,5 × (LSN), sauf pour les sujets recevant des anticoagulants dérivés de la coumarine ou un autre traitement anticoagulant similaire, qui doivent avoir un PT-INR dans la plage thérapeutique jugée appropriée par l’investigateur.
- Pour les femmes en âge de procréer : accord pour rester abstinent (s’abstenir de rapports hétérosexuels) ou utiliser des méthodes contraceptives, et accord pour ne pas donner d’ovules, tels que définis ci-dessous :
- Les femmes doivent rester abstinentes ou utiliser des méthodes contraceptives avec un taux d’échec de < 1 % par an pendant la période de traitement, 6 mois après la dernière dose de doxorubicine, 12 mois après la dernière dose de cyclophosphamide, 6 mois après la dernière dose de paclitaxel et 7 mois après la dernière dose de trastuzumab, de pertuzumab ou de T-DM1, selon la dernière éventualité. Les femmes doivent s’abstenir de donner des ovules pendant cette même période.
- Une femme est en âge de procréer si elle est post-ménarche, n’a pas atteint un état post-ménopausique (> 12 mois consécutifs d’aménorrhée sans cause identifiée autre que la ménopause) et n’a pas subi de stérilisation chirurgicale (ablation des ovaires et/ou de l’utérus). La définition du potentiel de procréation peut être adaptée pour s’aligner sur les directives ou les exigences locales.
- Parmi les exemples de méthodes contraceptives avec un taux d’échec de < 1 % par an, figurent la ligature bilatérale des trompes, la stérilisation masculine, les dispositifs intra utérins en cuivre, les contraceptifs hormonaux qui inhibent l’ovulation et les dispositifs intra-utérins libérant des hormones chez les femmes atteintes de tumeurs à récepteurs hormonaux négatifs uniquement ; l’utilisation de contraceptifs hormonaux et de dispositifs intra-utérins libérant des hormones est interdite chez les femmes atteintes de tumeurs à récepteurs hormonaux positifs.
- La fiabilité de l’abstinence sexuelle doit être évaluée en fonction de la durée de l’étude et du mode de vie préféré et habituel du patient. L’abstinence périodique (par exemple, les méthodes calendaires, d’ovulation, symptothermiques ou postovulantes) et le retrait ne sont pas des méthodes de contraception acceptables.
- Pour les hommes : accord de rester abstinent (s’abstenir de rapports hétérosexuels) ou d’utiliser des mesures contraceptives, et accord de ne pas donner de sperme, tels que définis ci dessous :
- Avec une partenaire féminine en âge de procréer qui n’est pas enceinte, les hommes qui ne sont pas chirurgicalement stériles doivent rester abstinents ou utiliser un préservatif plus une méthode contraceptive supplémentaire qui, ensemble, entraînent un taux d’échec de < 1 % par an pendant la période de traitement et pendant 6 mois après la dose finale de doxorubicine et/ou de cyclophosphamide, 6 mois après la dernière dose de paclitaxel et 7 mois après la dernière dose de trastuzumab, de pertuzumab ou de T-DM1, selon la dernière éventualité. Les hommes doivent s’abstenir de donner du sperme pendant cette même période. Les patients masculins sont encouragés à demander des conseils concernant la cryoconservation des spermatozoïdes avant de commencer le traitement à l’étude en raison de la possibilité d’infertilité avec la chimiothérapie.
- Avec une partenaire féminine enceinte, les hommes doivent rester abstinents ou utiliser un préservatif pendant la période de traitement et pendant 6 mois après la dernière dose de doxorubicine et/ou de cyclophosphamide, 6 mois après la dernière dose de paclitaxel et 7 mois après la dernière dose de trastuzumab, de pertuzumab ou de T-DM1, selon la dernière éventualité, pour éviter d’exposer l’embryon.
- La fiabilité de l’abstinence sexuelle doit être évaluée en fonction de la durée de l’étude et du mode de vie préféré et habituel du patient. L’abstinence périodique (par exemple, les méthodes calendaires, d’ovulation, symptothermiques ou postovulantes) et le retrait ne sont pas des méthodes de contraception acceptables.
Critère(s) de non-inclusion
- Cancer du sein de stade IV (métastatique).
- Hypersensibilité connue à l’un des excipients du trastuzumab, du pertuzumab, du carboplatine, du T-DM1, du docétaxel ou du paclitaxel.
- Patients atteints d’un cancer du sein invasif bilatéral synchrone.
- Traitement systémique antérieur pour le traitement du cancer du sein.
- Cancer du sein ulcéreux ou inflammatoire.
- A subi une biopsie incisionnelle et/ou excisionnelle de la tumeur primitive et/ou des ganglions lymphatiques axillaires.
- Procédure du ganglion sentinelle ou curage ganglionnaire axillaire avant le début du traitement néoadjuvant.
- Les patients ayant des antécédents de cancer du sein sont exclues. Les patients ayant des antécédents de tout autre cancer (à l’exception du cancer de la peau autre que le mélanome ou du carcinome in situ du col de l’utérus), à moins qu’ils ne soient en rémission complète sans traitement pendant au moins 3 ans, sont exclus. Pour les patients ayant des antécédents d’autres cancers autres que le sein dans les 3 ans et considérées comme présentant un faible risque de récidive selon le jugement de l’investigateur (par exemple, cancer papillaire de la thyroïde traité par chirurgie), l’éligibilité doit être discutée avec le promoteur de l’étude.
- Dysfonctionnement cardiorespiratoire tel que défini par l’un des éléments suivants avant la randomisation :
- Antécédents d’insuffisance cardiaque congestive de toute classification.
- Angine de poitrine nécessitant un médicament anti-angineux, arythmie cardiaque grave non contrôlée par un médicament adéquat, anomalie de conduction sévère ou maladie valvulaire cliniquement significative.
- Arythmies non contrôlées à haut risque (c.-à-d. tachycardie auriculaire avec une fréquence cardiaque > 100/min au repos, arythmie ventriculaire importante [tachycardie ventriculaire] ou bloc auriculo-ventriculaire [AV] de haut grade [bloc AV du deuxième degré de type 2 [Mobitz 2] ou bloc AV du troisième degré]).
- Symptômes significatifs (grade > 1) liés à une dysfonction ventriculaire gauche, à une arythmie cardiaque ou à une ischémie cardiaque.
- Infarctus du myocarde dans les 12 mois précédant la randomisation.
- Preuve d’infarctus transmural à l’ECG.
- Nécessité d’une oxygénothérapie.
- Dyspnée au repos.
- Intervention chirurgicale majeure, autre que pour le diagnostic, dans les 4 semaines précédant le début du traitement à l’étude ou l’anticipation de la nécessité d’une intervention chirurgicale majeure pendant l’étude.
- Infection grave dans les 4 semaines précédant le début du traitement à l’étude, y compris, mais sans s’y limiter, l’hospitalisation pour complications d’une infection, d’une bactériémie ou d’une pneumonie sévère.
- Antécédents de comorbidités importantes qui, de l’avis de l’investigateur, peuvent interférer avec la conduite de l’étude, l’évaluation de la réponse ou la procédure de consentement.
- Grossesse ou allaitement, ou intention de tomber enceinte pendant le traitement à l’étude ou dans les 6 mois suivant la dernière dose de paclitaxel, ou 7 mois après la dernière dose de trastuzumab, de pertuzumab ou de T-DM1, selon la dernière éventualité.
Remarque : Les femmes en âge de procréer doivent avoir un résultat négatif au test de grossesse sérique dans les 72 heures précédant le début du traitement à l’étude. - Les personnes privées de liberté ou placées sous garde protectrice ou sous tutelle.
- Patients ne voulant pas ou ne pouvant pas se conformer au suivi médical requis par l’essai pour des raisons géographiques, familiales, sociales ou psychologiques.
- Patients participant à d’autres essais cliniques / recevant d’autres traitements expérimentaux (IMPs) et leurs métabolites pertinents, ou ayant reçu des thérapies antérieures dont la toxicité pourrait (ou est susceptible de) se chevaucher avec celle du médicament expérimental et de ses métabolites pertinents, dans un délai correspondant à cinq fois la demi-vie du médicament / des métabolites (selon la durée la plus longue).
Calendrier prévisionnel
Lancement de l’étude : Novembre 2025
Fin estimée des inclusions : Novembre 2028
Nombre de patients à inclure : 304
Promoteur de l'étude
Fundacio Clinic Barcelona