NoLEEta
Retrait de la chimiothérapie dans le cancer du sein précoce à risque intermédiaire RH+ HER2-traité par Ribociclib (LEE011) en situation adjuvante, essai de phase III de non-infériorité
Type d'essai : Académique / Institutionnel
Etat de l'essai : Ouvert
Situation thérapeutique : Tumeur solide ( Adjuvant )
Etablissement(s) participant(s)
Dr Philippe FOLLANA
Pr Anthony GONCALVES
Dr Maya AL GHOR
Dr Julien GRENIER
Détails de l'essai
Objectif principal
Démontrer la non-infériorité d’un traitement adjuvant comprenant le ribociclib et la TE (bras A) par rapport au même traitement précédé d’une chimiothérapie adjuvante (bras B) dans le cas d’un cancer du sein à risque intermédiaire (RH+ HER2- EBC), en ce qui concerne la survie sans cancer du sein invasif (iBCFS).
Objectif(s) secondaire(s)
Evaluer l’efficacité des deux bras de traitement en fonction des paramètres suivants : Survie sans maladie invasive (iDFS), survie sans maladie à distance (DDFS) et taux de survie globale (SG) à 5, 8 et 10 ans et iBCFS par sous-groupes.
Evaluer le taux des différents types d’événements iBCFS dans chaque bras de traitement à 3, 5, 8 et 10 ans.
Evaluer la sécurité et la tolérabilité dans les deux bras de traitement en ce qui concerne la fréquence et la gravité des événements indésirables (EI).
Evaluer les changements de la qualité de vie liée à la santé dans les deux bras.
Résumé / schéma de l'étude
Après une chirurgie curative, les patients sont répartis de manière aléatoire en deux Bras :
• Bras expérimental : ribociclib et thérapie endocrinienne (TE) (bras A)
• Bras contrôle : chimiothérapie suivie de ribociclib et d’un traitement endocrinien (bras B)
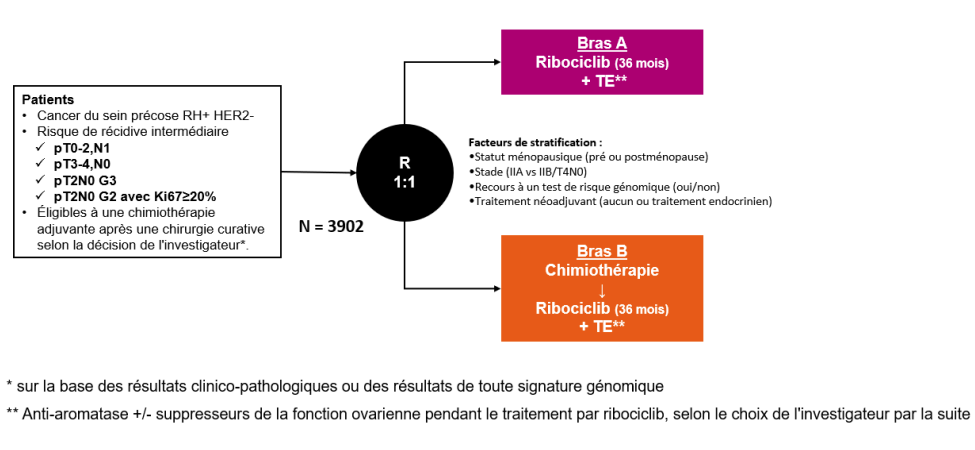
Critère(s) d'inclusion
- Le patient doit avoir signé un consentement éclairé écrit avant toute procédure de sélection spécifique à l’essai.
Note : Lorsque le patient est physiquement incapable de donner son consentement écrit, un témoin impartial de son choix, indépendant de l’investigateur ou du promoteur, peut confirmer par écrit le consentement du patient. - Le patient est âgé de ≥ 18 ans.
- La patiente est une femme dont le statut ménopausique est connu au moment de la randomisation.
Le statut post-ménopausique est défini comme suit :
a. La patiente a subi une ovariectomie bilatérale, ou
b. Âge ≥ 60 ans, ou
c. Âge < 60 ans et aménorrhée depuis 12 mois ou plus (en l’absence de chimiothérapie, de tamoxifène, de torémifène ou de suppression ovarienne) ou l’hormone folliculo-stimulante (FSH) et l’estradiol plasmatique se situent dans les plages postménopausiques selon les plages normales locales.
d. Si l’on prend du tamoxifène ou du torémifène et que l’on est âgé de moins de 60 ans, la FSH et le taux d’estradiol plasmatique doivent se situer dans l’intervalle postménopausique. - Les critères suivants doivent être remplis pour un carcinome mammaire invasif confirmé histologiquement, tel que déterminé par le pathologiste local :
a. Stade pathologique (8e édition de l’AJCC), y compris pT2 pN0 Grade 3 ou pT2 pN0 Grade 2 avec Ki67≥20%. ou pT0-2 pN1 ou pT3-4 pN0
b. RE-positif (avec des cellules tumorales présentant une coloration RE ≥10%) et HER2-négatif selon les directives ASCO/CAP les plus récentes.
Note : Les tumeurs multifocales et multicentriques sont autorisées si elles répondent aux critères du stade clinique II de la 8e édition de l’AJCC. Toutes les tumeurs doivent être RE positives et HER2-négatives. Les patientes atteintes d’un cancer du sein invasif bilatéral (diagnostiqué simultanément ou à moins de 6 mois d’intervalle) sont éligibles si toutes les lésions testées des deux côtés sont RE+ (c’est-à-dire ≥ 10 % de cellules positives) et HER2-, ET si une chirurgie adéquate a été réalisée dans les deux seins. - Éligible à la chimiothérapie sur décision de l’investigateur, sur la base des résultats clinicopathologiques ou des résultats d’une signature génomique.
- La patiente ne présente pas de contre-indication au traitement endocrinien adjuvant (TE) ou à la chimiothérapie dans le cadre de l’essai et il est prévu qu’elle soit traitée par TE pendant 5 ans (après la date de randomisation) ou plus.
- Une chirurgie curative pour la maladie invasive doit avoir été effectuée avec des marges chirurgicales négatives dans les 12 semaines précédant la randomisation. Si les marges chirurgicales sont positives, les patients sont éligibles si une reprise chirurgicale ou un autre traitement local adéquat (c’est-à-dire une radiothérapie locale) est prévu.
- Les femmes en âge de procréer doivent avoir un test de grossesse sérique (β-hCG) confirmé négatif avant de commencer le traitement à l’étude.
- Les femmes en âge de procréer doivent accepter d’utiliser une forme efficace de contraception pendant le traitement de l’étude et jusqu’à 21 jours après la dernière dose de médicaments de l’étude ou plus longtemps si nécessaire selon les normes de soins ;
- Statut de performance de l’Eastern Cooperative Oncology Group (ECOG) de 0 à 1 dans les 28 jours précédant la randomisation.
- Fonction hématologique, rénale et hépatique adéquate, comme indiqué ci-dessous :
a. Nombre absolu de neutrophiles (ANC) ≥ 1,5 x 109 /L
b. Numération plaquettaire ≥ 100 x 109 /L
c. Hémoglobine ≥ 9 g/dL
d. Bilirubine totale < LSN. Les patients présentant un syndrome de Gilbert connu peuvent être recrutés avec une bilirubine totale ≤3 x LSN ou une bilirubine directe ≤1,5 x LSN.
e. Alanine aminotransférase (ALT) et aspartate aminotransférase (AST) <2,5 x LSN
f. Créatinine sérique ≤1,5 mg/dL ou clairance de la créatinine calculée ≥60 mL/min/1.73m2 (équation CKD-EPI (2021))
g. Le potassium, le calcium total (corrigé en fonction de l’albumine sérique) et le magnésium doivent se situer dans les limites normales de l’établissement ou être corrigés pour se situer dans les limites normales à l’aide de suppléments avant la première dose du médicament à l’étude. - Valeurs standard de l’ECG à 12 dérivations évaluées, comme :
a. Intervalle QTcF (intervalle QT avec correction de Fridericia) au moment du dépistage < 450 millisecondes (msec)
b. Fréquence cardiaque au repos de 50 à 100 battements par minute (déterminée à partir de l’ECG) - Le patient doit être désireux et capable de se conformer aux visites programmées, aux plans de traitement, aux tests de laboratoire et aux autres procédures de l’essai.
- Absence de toute condition psychologique, familiale ou géographique susceptible d’entraver le respect du protocole de l’étude et du calendrier de suivi ; ces conditions doivent être discutées avec le patient avant l’inscription à l’essai.
- Les patients doivent être affiliés à un système de sécurité sociale (ou équivalent) conformément à la réglementation locale.
Critère(s) de non-inclusion
- Le patient a déjà reçu une chimiothérapie néoadjuvante pour son cancer du sein ou a déjà un inhibiteur de CDK4/6.
- Cancer du sein diagnostiqué alors que la patiente recevait du tamoxifène, du raloxifène ou des inhibiteurs de l’aromatase (IA) pour la réduction du risque (« chimioprévention ») de cancer du sein et/ou le traitement de l’ostéoporose au cours des 2 dernières années précédant la randomisation.
- Patient présentant une hypersensibilité connue à l’un des excipients du ribociclib et/ou de la TE (par exemple, problèmes héréditaires rares d’intolérance au galactose, déficit en lactase de Lapp, malabsorption du glucose-galactose et allergie au soja ou à l’arachide).
- Patiente présentant des preuves ou des antécédents de métastases à distance du cancer du sein au-delà des ganglions lymphatiques régionaux (stade IV selon la 8e édition de l’AJCC), de cancer du sein inflammatoire, de récidive du cancer du sein (locale ou à distance) ou d’un autre cancer du sein primaire.
- Le patient est atteint d’une tumeur maligne invasive concomitante ou d’une tumeur maligne invasive antérieure dont le traitement s’est achevé dans les deux ans précédant la randomisation.
Note : Les patients atteints d’un carcinome cutané basocellulaire ou spinocellulaire traité de manière adéquate ou d’un cancer du col de l’utérus in situ réséqué de manière curative sont éligibles. - Les patientes dont le cancer du sein est considéré comme insensible à la thérapie endocrinienne, selon l’avis de l’investigateur ; ceci peut inclure (mais n’est pas limité à) un cancer du sein classé comme » basal like » par les signatures moléculaires (si disponibles dans le dossier de la patiente) et/ou un cancer du sein avec une prolifération élevée persistante après une thérapie endocrinienne préopératoire.
- Le patient a subi une intervention chirurgicale majeure dans les 14 jours précédant le début du traitement de l’étude.
- Le patient a des antécédents connus d’infection par le virus de l’immunodéficience humaine (VIH) (le test n’est pas obligatoire) dont le traitement antirétroviral (ART) comporte un inhibiteur puissant du CYP3A4 connu avec un potentiel de DDI avec le ribociclib. Les patients séropositifs peuvent être recrutés s’ils remplissent les critères recommandés par les directives de la FDA et de l’ASCO (FDA Guidance, Uldrick et al. 2017) :
a. Numération des lymphocytes T CD4+ (CD4+) ≥ 350 cellules/µL, et
b. Pas d’antécédents d’infections opportunistes définissant le SIDA au cours des 12 derniers mois (les antimicrobiens prophylactiques sont autorisés s’il n’y a pas d’interactions médicamenteuses ou de toxicités se chevauchant), ET
c. Suivre un traitement antirétroviral qui n’est pas un inhibiteur puissant du CYP3A4 pendant au moins 4 semaines et avoir une charge virale VIH inférieure à 400 copies/ml avant l’inscription. Un traitement antirétroviral efficace est défini comme un médicament, une posologie et un calendrier associés à une réduction et à un contrôle de la charge virale. 9. - Le patient présente une infection active connue par le virus de l’hépatite B (VHB) ou le virus de l’hépatite C (VHC) (le test n’est pas obligatoire).
- Maladie cardiaque cliniquement significative et non contrôlée et/ou anomalie de repolarisation cardiaque, y compris l’un des éléments suivants :
a. Antécédents d’infarctus du myocarde (IM), d’angine de poitrine, de péricardite symptomatique ou de pontage aorto-coronarien dans les 6 mois précédant l’entrée dans l’essai.
b. Cardiomyopathie avérée.
c. Fraction d’éjection du ventricule gauche (FEVG) < 50 %, déterminée par une scintigraphie à acquisition multiple (MUGA) ou une échocardiographie (ECHO) (test non obligatoire).
d. Syndrome du QT long ou antécédents familiaux de mort subite idiopathique ou de syndrome du QT long congénital, ou l’un des éléments suivants :
• Facteurs de risque de torsades de pointes (TdP), notamment hypocalcémie, hypokaliémie ou hypomagnésémie non corrigées, antécédents d’insuffisance cardiaque ou antécédents de bradycardie cliniquement significative/symptomatique.
• Médicament(s) concomitant(s) présentant un risque connu d’allongement de l’intervalle QT et/ou connu(s) pour provoquer un TdP et ne pouvant être interrompu(s) ou remplacé(s) par un médicament alternatif sûr (par exemple dans les 5 demi-vies ou 7 jours précédant le début du traitement de l’essai).
• Impossibilité de déterminer l’intervalle QTcF.
e. Arythmies cardiaques cliniquement significatives (par exemple, tachycardie ventriculaire), bloc de branche gauche complet, bloc auriculo-ventriculaire (AV) de haut degré (par exemple, bloc bifasciculaire, bloc AV de type II de Mobitz et bloc AV du troisième degré).
f. Hypertension artérielle non contrôlée avec une pression artérielle systolique >160 mmHg. - Présence de toute autre condition médicale, y compris un dysfonctionnement respiratoire ou métabolique, des résultats d’examen physique ou de laboratoire qui font raisonnablement suspecter une contre-indication à l’utilisation d’un médicament expérimental, un impact potentiel sur le respect du protocole de l’étude, une influence sur l’interprétation des résultats ou un risque accru de complications du traitement pour les patients (tels qu’une dyspnée sévère au repos ou nécessitant une oxygénothérapie, des antécédents de résection chirurgicale majeure impliquant l’estomac ou l’intestin grêle, une maladie de Crohn ou une colite ulcéreuse préexistante, ou une affection chronique préexistante entraînant une diarrhée cliniquement significative).
- Antécédents de pneumopathie, quelle qu’en soit la cause.
- Le patient reçoit actuellement l’une des substances suivantes dans les 7 jours précédant la randomisation et qui ne peut être arrêtée dans les 7 jours précédant le début du traitement : a. Médicaments concomitants, suppléments à base de plantes et/ou fruits (pamplemousse, pomelos, carambole, oranges de Séville) et leurs jus qui sont connus comme étant de puissants inhibiteurs ou inducteurs du CYP3A4/5.
b. Médicaments dont la fenêtre thérapeutique est étroite et qui sont principalement métabolisés par le CYP3A4/5
c. Tout médicament interdit selon les instructions pour la goséréline, le leuprolide ou la triptoréline (patientes pré-ménopausées), l’anastrozole, l’exémestane, le létrozole ou le ribociclib.
d. Médicaments connus pour leur risque de prolonger l’intervalle QT ou de provoquer des torsades de pointes. - La patiente suit simultanément un traitement hormonal substitutif. L’œstrogénothérapie substitutive a été interrompue moins de deux semaines avant le début du traitement.
- Le patient reçoit actuellement ou a reçu des corticostéroïdes systémiques ≤ 2 semaines avant le début du traitement de l’essai, ou n’a pas complètement récupéré des effets secondaires d’un tel traitement.
Note : Les utilisations suivantes de corticostéroïdes sont autorisées : une courte durée (<5 jours) de corticostéroïdes systémiques ; toute durée d’applications topiques (par exemple pour les éruptions cutanées), de pulvérisations inhalées (par exemple pour les maladies obstructives des voies respiratoires), de gouttes pour les yeux ou d’injections locales (par exemple intraarticulaires). - Le patient présente une autre pathologie grave et/ou non contrôlée qui, selon l’investigateur, entraînerait des risques inacceptables pour la sécurité, contre-indiquerait la participation du patient à l’essai clinique ou compromettrait le respect du protocole (par exemple, pancréatite chronique, hépatite chronique active, cirrhose du foie ou toute autre maladie hépatique importante, infections fongiques, bactériennes ou virales actives non traitées ou non contrôlées, infection active nécessitant un traitement antibactérien systémique, etc.) ou limiterait l’espérance de vie à ≤5 ans.
- Participation à d’autres études impliquant un ou des médicaments expérimentaux dans les 30 jours précédant la randomisation ou dans les 5 demi-vies du ou des médicaments expérimentaux (le délai le plus long étant retenu), ou participation à tout autre type de recherche médicale jugée scientifiquement ou médicalement incompatible avec cet essai. Si le patient est inscrit ou prévoit d’être inscrit dans une autre étude n’impliquant pas de médicament expérimental, l’accord du promoteur est nécessaire pour établir l’éligibilité.
- Incapacité ou refus d’avaler des pilules orales.
- Présence d’un syndrome de malabsorption ou de toute autre affection susceptible d’entraver l’absorption des médicaments à l’étude dans le tractus gastro-intestinal.
- Tout facteur psychologique, familial, sociologique ou géographique susceptible d’entraver l’adhésion au protocole de l’étude et au calendrier de suivi.
- Femmes enceintes ou allaitantes ou femmes qui prévoient d’être enceintes ou d’allaiter pendant les 48 premiers mois du traitement adjuvant.
- Personnes privées de liberté ou placées sous protection ou tutelle.
Calendrier prévisionnel
Lancement de l’étude : Décembre 2025
Fin estimée des inclusions : Novembre 2033
Nombre de patients à inclure : 3902
Coordonnateur de l'étude
Pr François-Clément BIDARD – Institut Curie – CLCC Villejuif
Promoteur de l'étude
UNICANCER