SAFIR 03 LibHERTy
Programme de dépistage d’ADN tumoral (ADNct) circulant chez des patient(e)s présentant un cancer du sein métastatique positif aux récepteurs hormonaux (RH+) , HER2 faible pour la détection des patients à haut risque de récidive sous traitement par un inhibiteur de CDK4/6 et hormonothérapie, suivi d’une étude de phase II avec du trastuzumab-deruxtecan chez les patients ayant de l’ADNct persistant après 1 mois de ce traitement
Type d'essai : Académique / Institutionnel
Etat de l'essai : Ouvert
Situation thérapeutique : Tumeur solide ( Localement avancée / Non résécable , Métastatique / Rechute )
Etablissement(s) participant(s)
Dr Bertrand BILLEMONT
Détails de l'essai
Objectif principal
Evaluer si le passage précoce du traitement standard (à 8 semaines) par une hormonothérapie (IAou fulvestrant) associé à un inhibiteur de CDK4/6, au trastuzumab deruxtecan (T-DXd) pour une population dont on s’attend à ce qu’elle développe une résistance au traitement standard d’après la réponse moléculaire, est associé à une amélioration de la survie sans progression (PFS).
Objectif(s) secondaire(s)
Evaluer et comparer la survie globale (OS).
Evaluer les taux de réponse objective (ORR).
Evaluer la durée de la réponse (DoR).
Evaluer le taux de bénéfice clinique (CBR).
Evaluer le délai de réponse (TTR).
Evaluer la sécurité du T-DXd.
Comparer la PFS et l’OS du passage précoce (à 8 semaines) d’un traitement hormonal (IA ou fulvestrant) associé à des inhibiteurs CDK4/6 inh au T-DXd, par rapport à la poursuite d’une hormonothérapie associée à un inhibiteur CDK4/6 avec les données d’essais historiques (PADA-1 et Destiny Breast-06).
Résumé / schéma de l'étude
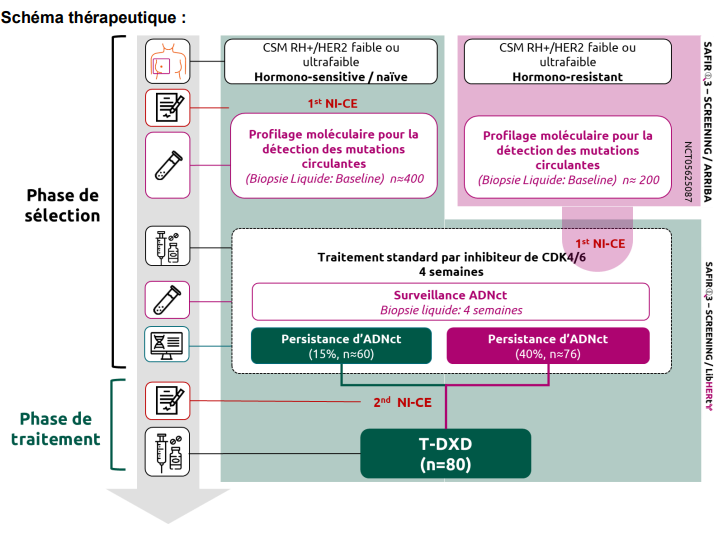
Critère(s) d'inclusion
Phase de sélection
- Le patient doit avoir signé un consentement éclairé écrit de la phase de sélection avant toute procédure spécifique à l’étude.
Note : Lorsque le patient est physiquement incapable de donner son consentement écrit, une personne de confiance de son choix, indépendante de l’investigateur ou du promoteur, peut confirmer par écrit le consentement du patient. - Le patient est âgé de ≥ 18 ans.
- Cancer du sein documenté :
- Métastatique et peut faire l’objet d’une biopsie en vue d’une confirmation histologique ou cytologique ultérieure.
- HER2 faible (HER2 1+, ou 2+ et ISH négatif) ou HER2 ultra faible (IHC 0 avec coloration membranaire incomplète et faible dans >0 et ≤ 10% des cellules tumorales) sur le matériel tumoral disponible le plus récent, tel que défini par le pathologiste local selon les directives ASCO/CAP.
- Récepteurs hormonaux positifs (RH+), définis comme ayant une expression du récepteur aux œstrogènes (RE) et/ou du récepteur à la progestérone (RP) dans ≥10 % des noyaux des cellules tumorales immunoréactives, en situation métastatique.
- Le patient présente soit :
- Une rechute métastatique pendant ou dans l’année qui suit la fin du traitement hormonal adjuvant (résistant à l’IA),
ou - une rechute métastatique, localisée au poumon et/ou au foie et/ou à d’autres localisations viscérales, plus d’un an après la fin du traitement adjuvant aux IA ou des métastases de novo AI.
- Une rechute métastatique pendant ou dans l’année qui suit la fin du traitement hormonal adjuvant (résistant à l’IA),
- Pour la population résistante à l’IA : le patient a déjà débuté la phase de dépistage de l’ADNct de l’étude SAFIR 03 – SCREENING/ARRIBA et a respecté tous les critères d’inclusion et d’exclusion.
- Le patient n’a reçu aucune thérapie en situation métastatique.
- Le patient est éligible à un traitement de première ligne par un inhibiteur de CDK4/6 commercialisé (palbociclib, ribociclib ou abemaciclib) en association avec un IA ou du fulvestrant, conformément à son autorisation de mise sur le marché.
- L’échelle de statut de performance ECOG du patient est ≤ 1.
- Le patient a une fonction hématologique et de ses organes adéquate.
- Le patient présente une maladie mesurable ou évaluable selon RECIST v1.1.
- Disponibilité d’un échantillon de tumeur métastatique archivé (FFPE) pour la recherche exploratoire. Les métastases osseuses sont acceptées si le tissu est représentatif du tissu tumoral (au moins 10 % de cellularité tumorale).
- Le patient doit être disposé et capable de se conformer au protocole pendant toute la durée de l’étude, y compris aux visites prévues, au plan de traitement, aux tests de laboratoire et aux autres procédures de l’étude.
- Le patient doit être affilié à un régime de sécurité sociale (ou équivalent).
Phase de traitement
- Le patient doit avoir signé un consentement éclairé écrit de la phase de traitement avant toute procédure spécifique à l’étude.
Note : Lorsque le patient est physiquement incapable de donner son consentement écrit, une personne de confiance de son choix, indépendante de l’investigateur ou du promoteur, peut confirmer par écrit le consentement du patient. - Le patient doit avoir arrêté l’inhibiteur CDK4/6 (palbociclib, ribociclib ou abemaciclib) au moins 7 jours avant la randomisation, mais pas plus de 14 jours.
- Le patient doit présenter une absence de chute de l’ADNct déterminée par un dosage de l’ADNct après 4 semaines de traitement standard par un inhibiteur de CDK4/6. Une absence de chute est définie par une réponse moléculaire (MR) ≥ 0,5 [Zhang 2020].
- L’échelle de statut de performance ECOG du patient est ≤ 1.
- Fraction d’éjection du ventricule gauche (FEVG) ≥ 50 % dans les 28 jours précédant l’inclusion.
- Le patient a une fonction hématologique et de ses organes adéquate, dans les 14 jours précédant l’inclusion telles que définies par les valeurs de laboratoire suivantes :
- Numération absolue des neutrophiles ≥ 1500/mm3
- Numération des plaquettes ≥ 100 000/mm3
- Hémoglobine ≥ 9,0 g/dL
- Créatinine sérique ≤ 1,5 × LSN ou clairance de la créatinine ≥ 30 mL/min
- Albumine sérique ≥ 2.5 g/dL
- Bilirubine totale ≤ 1,5× LIN (<3 LSN en cas de maladie de Gilbert documentée)
- En l’absence de métastases hépatiques, alanine aminotransférase (ALT) et aspartate aminotransférase (AST) ≤3 × LSN. Si le participant présente des métastases hépatiques et que les taux d’ALT et d’AST sont < 5 × LSN, il sera éligible pour l’étude.
- Fonction de coagulation sanguine adéquate : définie par un rapport international normalisé/temps de prothrombine ≤ 1,5 × LSN et un temps de thromboplastine partielle ou un temps de thromboplastine partielle activée dans les limites de la normale.
Note : La transfusion (de globules rouges ou de plaquettes) ou l’administration de G-CSF n’est pas autorisée dans les 14 jours précédant le jour où la fonction de la moelle osseuse
est évaluée, ni à tout moment après ce jour et avant le C1D1.
- Les femmes en âge de procréer doivent avoir un résultat négatif au test de grossesse sérique (avec une sensibilité d’au moins 25 mUI/mL) dans les 3 jours précédant l’inclusion.
- Les hommes ou les femmes en âge de procréer doivent accepter d’utiliser un moyen de contraception efficace pendant toute la durée de l’étude et pendant au moins 7 mois après la dernière dose du traitement à l’étude pour les femmes, et de 4 mois pour les hommes.
- Le patient doit être disposé et capable de se conformer au protocole pendant toute la durée de l’étude, y compris aux visites prévues, au plan de traitement, aux tests de laboratoire et aux autres procédures de l’étude.
Critère(s) de non-inclusion
Phase de sélection
- Le patient est éligible à la chimiothérapie en raison d’une crise viscérale, (dysfonctionnement sévère des organes, évalué par les signes et symptômes, les analyses de biologie médicale et la progression rapide de la maladie).
Note : La crise viscérale n’est pas la simple présence de métastases viscérales mais implique une importante atteinte d’organe conduisant à une indication clinique pour le traitement le plus rapidement efficace. - Le patient est atteint d’un cancer du sein pouvant faire l’objet d’une résection ou d’une radiothérapie à visée curative.
- Exposition antérieure à anticorps conjugué (ADC) ou à un inhibiteur de CDK4/6 (en situation métastatique). L’inhibiteur de CDK4/6 administré en situation adjuvante doit être arrêté depuis au moins 12 mois avant la sélection.
- Le patient a déjà initié le traitement par inhibiteur de CDK4/6,
- Le patient est incapable d’avaler des comprimés.
- Le patient a des antécédents de pneumopathie interstitielle (non infectieuse) nécessitant des stéroïdes, présente une pneumopathie interstitielle en cours ou une pneumopathie interstitielle présumée ne peut être exclue par l’imagerie au moment de l’examen initial.
- Le patient a des antécédents de réactions d’hypersensibilité graves aux substances médicamenteuses ou aux ingrédients inactifs des traitements.
- Le patient a des antécédents de réactions d’hypersensibilité graves à d’autres anticorps monoclonaux.
- Le patient est privé de liberté ou sous tutelle.
- Facteurs sociaux, familiaux ou géographiques susceptibles d’entraver la participation à l’étude ou le suivi.
Phase de traitement
- Le patient est éligible à la chimiothérapie en raison d’une crise viscérale, (dysfonctionnement sévère des organes, évalué par les signes et symptômes, les analyses de biologie médicale et la progression rapide de la maladie).
Note : La crise viscérale n’est pas la simple présence de métastases viscérales mais implique une importante atteinte d’organe conduisant à une indication clinique pour le traitement le plus rapidement efficace. - Le patient est résistant à l’IA et est éligible à l’essai SAFIR 03 – ARRIBA (patient muté PIK3CA), sauf si l’étude SAFIR 03 – ARRIBA atteint la fin des inclusions ou est arrêtée par décision du comité de pilotage de l’étude.
- Le patient a reçu plus de 2 cycles de l’inhibiteur de CDK4/6 en cours, associé aux IA ou au fulvestrant.
- Le patient a interrompu le traitement par inhibiteur de CDK4/6 pendant plus de 14 jours.
- Le patient présente des signes de progression clinique ou radiologique de la maladie.
- Le patient présente des effets indésirables non résolus d’un traitement anticancéreux antérieur, définis comme des toxicités (autres que l’alopécie) encore non résolus à un grade ≤ 1 ou à la baseline.
Note : Les patients peuvent être recrutés en cas de toxicité chronique stable de grade 2 (définie par l’absence d’aggravation à > grade 2 pendant au moins 3 mois avant le recrutement et prise en charge par un traitement standard) que l’investigateur juge liée à un traitement anticancéreux antérieur, comme par exemple la neuropathie ou des fatigues induites par la chimiothérapie. - Le patient a eu une tumeur maligne dans les 3 ans, autre que celle à l’étude, à l’exception : du cancer de la peau non-mélanome réséqué de manière adéquate, une maladie in situ traitée de manière curative, d’autres tumeurs solides traitées de manière curative, ou un cancer du sein controlatéral.
- Le patient est considéré comme présentant un risque médical élevé en raison d’une maladie systémique grave ou non contrôlée, y compris : un diabète sucré, une maladie pulmonaire cliniquement significative, un trouble neurologique cliniquement significatif, une pancréatite chronique, une infection chronique active par le virus de l’hépatite B, le virus de l’hépatite C ou le VIH, des infections fongiques, bactériennes ou virales actives non traitées ou non contrôlées, une immunodéficience primaire active, etc.
- Maladie cardiovasculaire importante ou non contrôlée, y compris l’une des maladies suivantes :
- Antécédents d’infarctus du myocarde dans les 6 mois précédant l’inclusion,
- Antécédents d’insuffisance cardiaque congestive symptomatique (classe II à IV de la New York Heart Association),
- Patient présentant un allongement de l’intervalle QT corrigé (QTc) >470 ms (femmes) ou >450 ms (hommes) sur la base de la moyenne de l’ECG à 12 dérivations réalisé en triplicat lors de de la visite de sélection,
- Les patients dont le taux de troponine est supérieur à la limite supérieure de la normale (LSN) au moment de la sélection (tel que défini par le fabricant) et qui ne présentent aucun symptôme lié au myocarde doivent faire l’objet d’une consultation cardiologique avant d’être recrutés afin d’exclure un infarctus du myocarde.
- Atteinte pulmonaire cliniquement grave résultant de maladies pulmonaires actuelles, y compris, mais sans s’y limiter, tout trouble pulmonaire sous-jacent (par ex, embolie pulmonaire dans les trois mois précédant le début de l’étude, asthme sévère, bronchopneumopathie chronique obstructive (BPCO) sévère, maladie pulmonaire restrictive, épanchement pleural, etc.), et toute maladie auto-immune, du tissu conjonctif ou inflammatoire avec atteinte pulmonaire (par exemple, polyarthrite rhumatoïde, syndrome de Sjögren, sarcoïdose, etc.
- Le patient présente une compression de la moelle épinière ou des métastases du système nerveux central cliniquement actives, définies comme non traitées ou symptomatiques, ou nécessitant un traitement par corticostéroïdes ou anticonvulsivants pour contrôler les symptômes associés.
- Intervention chirurgicale majeure dans les 4 semaines précédant l’inclusion.
- Le patient a reçu une radiothérapie dans les 4 semaines ou une irradiation à champ limité palliative dans les 2 semaines avant de commencer le médicament de l’étude, ou n’a pas récupéré au grade 1 ou mieux des effets secondaires liés à cette thérapie (les exceptions incluent l’alopécie) et/ou chez qui ≥ 25% de la moelle osseuse a été irradiée.
- Patient utilisant des médicaments susceptibles d’avoir une interaction pharmacocinétique avec les médicaments expérimentaux. L’utilisation concomitante d’inhibiteurs puissants du CYP3A4 ou d’inhibiteurs de l’OATP 1B doit être évitée. Si l’utilisation concomitante d’inhibiteurs puissants du CYP3A4 ou de l’OATP 1B est inévitable, il faut envisager de retarder le traitement par trastuzumab-deruxtecan jusqu’à ce que les inhibiteurs aient disparu de la circulation (environ 3 demi-vies d’élimination des inhibiteurs) lorsque cela est possible. Si un inhibiteur puissant du CYP3A4 ou un inhibiteur de l’OATP 1B est coadministré et que le traitement par T-DXd ne peut pas être retardé, les patients doivent être étroitement surveillés pour détecter les réactions indésirables.
- Patient recevant un médicament susceptible de provoquer un allongement de l’intervalle QTc ou une arythmie cardiaque. Le pimozide (Orap®) et le cisapride (Prepulsid®) sont strictement contre-indiqués : ils sont associés à un risque majeur de trouble du rythme ventriculaire.
- Administration d’un vaccin vivant atténué (les vaccins adénoviraux à ARNm et à réplication déficiente ne sont pas considérés comme des vaccins vivants atténués) dans les 30 jours précédant la première dose de trastuzumab deruxtecan. Remarque : les patients, s’ils sont recrutés, ne doivent pas recevoir de vaccin vivant pendant l’étude et jusqu’à 30 jours après la
dernière dose d’IMP. - Participation à une étude clinique thérapeutique dans les 4 semaines précédant l’inclusion.
- La patiente est actuellement enceinte, allaite ou envisage de le devenir. 19. Le patient a des antécédents de toxicomanie ou tout autre état pathologique, tel que des troubles cardiaques ou psychologiques cliniquement significatifs, qui pourraient, de l’avis de l’investigateur, interférer avec la participation du patient à l’étude clinique ou avec l’évaluation des résultats de l’étude clinique.
- Le patient est privé de liberté ou sous tutelle.
- Facteurs sociaux, familiaux ou géographiques susceptibles d’entraver la participation à l’étude ou le suivi.
Calendrier prévisionnel
Lancement de l’étude : Juillet 2025
Fin estimée des inclusions : Juillet 2028
Nombre de patients à inclure : Phase de sélection : environ 600 patients. Phase de traitement : 80 patients.
Coordonnateur de l'étude
Pr Fabrice André – Gustave Roussy – CLCC Villejuif
Promoteur de l'étude
UNICANCER