OPT-PEMBRO
OPTimiser la prescription adjuvante de PEMBROlizumab chez les patients atteints d’un cancer du sein triple négatif au stade précoce ayant obtenu une réponse pathologique complète après une chimiothérapie néoadjuvante standard et le pembrolizumab
Phase : III
Type d'essai : Académique / Institutionnel
Etat de l'essai : Ouvert
Situation thérapeutique : Tumeur solide ( Adjuvant )
Type d'essai : Académique / Institutionnel
Etat de l'essai : Ouvert
Situation thérapeutique : Tumeur solide ( Adjuvant )
Etablissement(s) participant(s)
Dr Julien GRENIER
Détails de l'essai
Objectif principal
Démontrer la non-infériorité de la surveillance active par rapport à 6 mois (9 cycles) de pembrolizumab adjuvant après la chirurgie en termes de survie sans événement chez les patients ayant obtenu une pRC après une chimiothérapie néoadjuvante associé à du pembrolizumab.
Objectif(s) secondaire(s)
Sécurité et tolérance.
Qualité de vie.
Fertilité.
Efficacité.
Résumé / schéma de l'étude
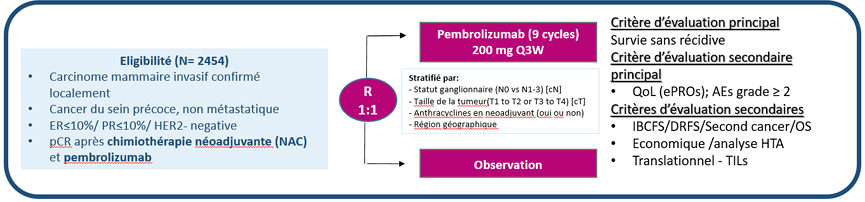
Critère(s) d'inclusion
- Le patient doit avoir signé un consentement éclairé écrit avant toute procédure liée à l’essai. Lorsque le patient est physiquement incapable de donner son consentement écrit, une personne de confiance de son choix, indépendante de l’investigateur ou du promoteur, peut confirmer par écrit le consentement du patient.
- Âge ≥ 18 ans.
- Statut de performance de l’Eastern Cooperative Oncology Group (ECOG) de 0 à 2.
- Cancer du sein de stade T1cN1-2 ou T2-4N0-2 triple négatif ou ER/PR faible (récepteurs d’œstrogènes (ER) et de progestérone (PR) ≤10% ; HER2 négatif selon les directives ASCO/CAP), histologiquement documenté. Stadification selon les critères anatomiques de stadification de la tumeur primaire et des ganglions lymphatiques régionaux de l’American Joint Committee on Cancer (AJCC), 8e édition, tels que déterminés par l’investigateur lors de l’évaluation radiologique, clinique ou les deux.
- Patients précédemment traités par chimiothérapie néoadjuvante en association avec le pembrolizumab pour un minimum de 6 cycles (Toute la chimiothérapie systémique doit avoir été achevée en préopératoire).
- Absence de maladie invasive résiduelle dans le sein ou les ganglions lymphatiques après l’achèvement du traitement néoadjuvant (c’est-à-dire ypT0 ypN0 dans le système de stadification actuel de l’AJCC) (un carcinome canalaire in situ [CCIS] résiduel est autorisé).
- Avoir subi une exérèse adéquate du cancer du sein (ablation chirurgicale de toute la maladie cliniquement évidente dans le sein et les ganglions lymphatiques) :
- Chirurgie mammaire : les patientes doivent avoir subi une chirurgie conservatrice du sein ou une mastectomie totale avec des marges histologiquement négatives pour les tumeurs invasives et le DCIS. Les patientes dont les marges sont positives pour le carcinome lobulaire in situ (LCIS) sont éligibles sans résection supplémentaire.
- Chirurgie des ganglions lymphatiques : les patientes doivent avoir subi une biopsie du ganglion lymphatique sentinelle (SLNB) et/ou une dissection du ganglion lymphatique axillaire (ALND) pour évaluer le statut pathologique du ganglion.
- Patients ayant reçu une radiothérapie locorégionale adéquate ou ayant prévu une radiothérapie locorégionale adéquate.
- Les fonctions des organes et de la moelle osseuse sont adéquates. Tous les tests de laboratoire de dépistage doivent être effectués dans les 28 jours précédant la randomisation :
- Nombre absolu de neutrophiles (ANC) ≥ 1 000 /µL
- Plaquettes ≥ 100 000 /µL
- Hémoglobine ≥ 9 g/dL
- Clairance de la créatinine ≥ 30 ml/min pour les patients dont le taux de créatinine est supérieur à 1,5 fois x LSN de l’institution.
- Bilirubine totale ≤ 1,5 x LSN ou bilirubine directe ≤ LSN pour les patients dont le taux de bilirubine totale est > 1,5 LSN (les patients atteints de la maladie de Gilbert avec une bilirubine totale ≤ 2,5 x LSN et une bilirubine directe dans les limites normales sont autorisés).
- Aspartate aminotransférase (ASAT) et alanine aminotransférase (ALAT) ≤ 2,5 x LSN.
- La randomisation doit avoir lieu au maximum 12 semaines après la chirurgie mammaire. La radiothérapie adjuvante est autorisée. Si elle est administrée, à la discrétion de l’investigateur, elle peut être administrée en même temps que le pembrolizumab.
- Les patientes ne doivent pas être enceintes ou allaitantes (pour les femmes en âge de procréer uniquement, un test de grossesse sanguin négatif doit être obtenu dans les 7 jours avant le jour 1 du cycle 1).
- Les femmes en âge de procréer et les patients de sexe masculin doivent accepter d’utiliser une forme efficace de contraception jusqu’à 4 mois après la dernière dose des médicaments de l’étude.
- Les patients doivent être capables et désireux de se conformer aux visites et aux procédures de l’étude conformément au protocole.
- Les patients doivent être affiliés à un système de sécurité sociale (ou équivalent).
Critère(s) de non-inclusion
- Preuve radiologique ou clinique d’une maladie métastatique (stade IV) documentée par imagerie ou examen clinique.
- Preuve de la récurrence de la maladie après un traitement préopératoire et une intervention chirurgicale.
- Tout antécédent de cancer du sein invasif (ipsi- ou controlatéral).
- Patients ayant des antécédents d’une autre tumeur maligne sans rémission complète depuis plus de 5 ans, à l’exception du carcinome cervical in situ correctement traité et du cancer de la peau sans mélanome.
- Les patients pour lesquels le pembrolizumab a été définitivement interrompu pendant la phase néoadjuvante du traitement en raison d’un EI lié au pembrolizumab.
- Antécédents d’intolérance, y compris réaction à la perfusion de grade 3 ou 4, ou hypersensibilité au pembrolizumab, aux protéines murines ou à tout composant du produit.
- Conditions médicales nécessitant des stéroïdes systémiques chroniques (>10 mg de prednisone ou équivalent) ou toute autre forme de médicament immunosuppresseur au cours des 2 dernières années. La thérapie de remplacement (par exemple, la thyroxine, l’insuline, la thérapie de remplacement des corticostéroïdes physiologiques en cas d’insuffisance surrénale ou hypophysaire) n’est pas considérée comme une forme de traitement systémique.
- Maladie hépatique active connue, par exemple due au VHB, au VHC, à des troubles hépatiques auto-immuns ou à une cholangite sclérosante.
- Patients infectés par le VIH sous traitement antirétroviral efficace avec une charge virale détectable dans les 6 mois précédant l’inscription.
- Les patients ayant des antécédents connus ou des symptômes actuels de maladie cardiaque, ou des antécédents de traitement par des agents cardiotoxiques, doivent faire l’objet d’une évaluation du risque clinique de la fonction cardiaque à l’aide de la classification fonctionnelle de la New York Heart Association. Pour être éligibles à cet essai, les patients doivent être en classe 2B ou mieux.
- Les patients qui ne veulent pas ou ne peuvent pas se conformer au suivi médical requis par l’essai pour des raisons géographiques, familiales, sociales ou psychologiques.
- Les personnes privées de liberté ou placées sous protection ou tutelle.
- Participation à un autre essai thérapeutique dans les 30 jours précédant la randomisation.
Calendrier prévisionnel
Lancement de l’étude : Août 2025
Fin estimée des inclusions : Août 2029
Nombre de patients à inclure : 2454
Coordonnateur de l'étude
Dr Joana MOURATO RIBEIRO – Gustave Roussy – CLCC Villejuif
Promoteur de l'étude
UNICANCER
Dernière mise à jour le 16 février 2026