PRODIGE 115 – FFCD 2406 - ULYSSE
FOLFOX/FOLFIRI + Bévacizumab ou Fruquintinib en 2ème ligne du Cancer Colorectal métastatique (CCRm) : essai de phase II randomisée non comparative
Phase : II
Type d'essai : Académique / Institutionnel
Etat de l'essai : Ouvert
Situation thérapeutique : Tumeur solide ( Métastatique / Rechute , Localement avancée / Non résécable )
Type d'essai : Académique / Institutionnel
Etat de l'essai : Ouvert
Situation thérapeutique : Tumeur solide ( Métastatique / Rechute , Localement avancée / Non résécable )
Etablissement(s) participant(s)
Pr Laetitia DAHAN
Détails de l'essai
Objectif principal
Taux de contrôle de la maladie (DCR) évalué 4 mois après le début du traitement.
Objectif(s) secondaire(s)
Tolérance et sécurité du traitement.
Survie sans progression (SSP) selon RECIST v1.1, évaluée par l’investigateur.
Taux de réponse objective (ORR) selon RECIST v1.1, évalué par l’investigateur.
Durée de la réponse (DOR) selon RECIST v1.1, évaluée par l’investigateur.
Survie globale (SG).
Qualité de vie (QLQ-C30).
Délai de dégradation de l’OMS > 2.
Résumé / schéma de l'étude
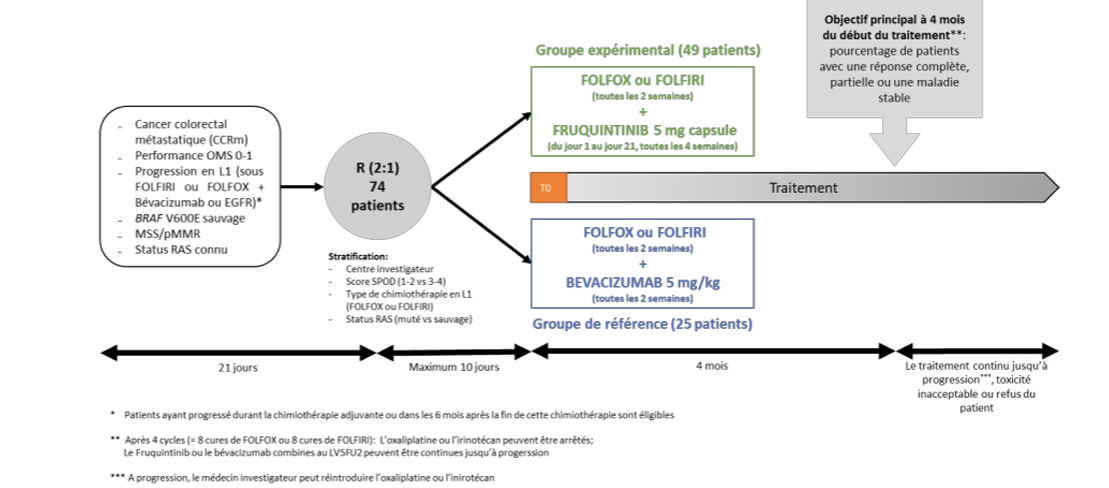
Critère(s) d'inclusion
- Âge ≥ 18 ans et ≤ 80 ans ; à condition que le score du questionnaire gériatrique G8 soit >14 pour les patients âgés de 75 ans ou plus.
- Patients ayant un diagnostic de CCRm confirmé histologiquement, avec une progression documentée de la maladie (selon RECIST v1.1, évaluée par l’investigateur et confirmée par TDM ou IRM).
- Patients ayant reçu un traitement de première intention par Bévacizumab ou un EGFRi, en association avec FOLFOX ou FOLFIRI, pour un CCRm non résécable. Les patients qui ont progressé pendant la chimiothérapie adjuvante (FOLFOX) ou dans les 6 mois suivant son achèvement sont éligibles à l’inclusion.
- Tumeur non résécable au moment de l’inclusion.
- Patients présentant au moins une lésion métastatique mesurable/évaluable selon les critères RECIST v1.1 ; les images doivent être disponibles pour être collectées.
- Métastases non accessibles à la chirurgie et/ou à la thermo-ablation et/ou à la radiothérapie stéréotaxique.
- Tumeur BRAF V600E sauvage.
- Tumeur MSS/pMMR.
- Statut de performance de l’OMS 0 ou 1.
- Paramètres disponibles pour calculer le score SPOD : OMS, hémoglobine, numération plaquettaire, rapport globules blancs/nucléaires absolus, lactate déshydrogénase (LDH), phosphatase alcaline et nombre de sites métastatiques.
- Fonctions hépatiques adéquates : Bilirubinémie totale < 1.5 LSN, ASAT et ALAT ≤ 3 LSN.
- Fonctions hématologiques adéquates (hémoglobine ≥10g/dL, plaquettes ≥100G/L, neutrophiles ≥1,5G/L) et fonctions rénales adéquates (clairance de la créatinine ≥ 50 ml/min selon CKD-EPI) adéquates.
- Protéinurie sur bandelette urinaire < 2+ ; si 2+ ou plus, la protéinurie doit être ≤1g/24h.
- Espérance de vie ≥ 3 mois.
- Les femmes en âge de procréer doivent accepter d’utiliser une méthode de contraception hautement efficace pendant l’essai et pendant au moins 15 mois après l’arrêt des traitements expérimentaux. Les hommes qui ont des relations sexuelles avec des femmes en âge de procréer doivent accepter d’utiliser une méthode de contraception hautement efficace pendant le traitement et pendant au moins 12 mois après l’arrêt des traitements expérimentaux.
- Capacité du patient à comprendre la note d’information, et de signer/dater le formulaire de consentement éclairé avant toute procédure spécifique à l’étude.
- Patient affilié à un régime de sécurité sociale.
- Échantillon de tumeur et compte rendu anatomopathologique disponibles pour être collectés.
Critère(s) de non-inclusion
- Patients ayant reçu plus d’un traitement systémique antérieur.
- FOLFIRINOX +/- thérapie ciblée en première ligne.
- Statut RAS inconnu.
- Métastases cérébrales connues.
- Carcinomatose péritonéale connue s’il y a des signes d’occlusion ou de sous-occlusion clinique.
- Antécédents d’ulcération gastrique, ou infarctus du myocarde, ou coronaropathie sévère ou dysfonctionnement cardiaque sévère, au cours des 6 derniers mois précédant le début du traitement.
- Patients présentant un déficit en dihydropyrimidine déshydrogénase (uracilémie ≥ 16 ng/mL).
- Hypersensibilité à l’un des médicaments de l’étude ou à l’un de ses excipients.
- Incapacité d’avaler les gélules.
- Vaccins vivants atténués 30 jours avant le début du traitement.
- Fracture osseuse non traitée.
- Diathèse hémorragique ou coagulopathie significative (en l’absence de traitement anticoagulant).
- Chirurgie majeure, biopsie ouverte ou lésion traumatique majeure dans les 30 jours précédents ou nécessité d’une chirurgie majeure pendant l’essai.
- Femmes enceintes ou allaitantes ou patients n’ayant pas de contraception adéquate.
- Déficit connu en Uridine Diphosphate Glucuronyltransférase (UGT1A1) ou maladie de Gilbert connue.
- Inducteurs puissants du CYP3A4 (traitement au millepertuis (Hypericum perforatum), à la fampicine, au phénobarbital, à la primidone, à la phénytoïne et à la carbamazépine).
- Inhibiteurs puissants du CYP3A4, utilisation continue d’antifongiques azolés (posaconazole, voriconazole, itraconazole, isavuconazole), ritonavir, vérapamil, diltiazem, jus de pamplemousse (équivalent à un demi-pamplemousse frais/jour).
- Traitement concomitant ou récent par la sorivudine ou ses analogues (y compris la brivudine) dans les 4 semaines précédant l’administration du traitement prévu par le protocole (lié au fluorouracile).
- Selon le RCP du Bévacizumab : hypersensibilité aux produits à base de cellules ovariennes de hamster chinois (CHO) ou à d’autres anticorps recombinants humains ou humanisés ; perforation gastrointestinale.
- Selon le RCP de l’irinotécan, en cas de maladie inflammatoire chronique de l’intestin et/ou d’occlusion intestinale.
- Selon le RMP de l’oxaliplatine : en cas de neuropathie sensorielle périphérique avec déficience fonctionnelle avant le premier traitement, en cas d’hypokaliémie, d’hypomagnésémie ou d’hypocalcémie, compte tenu de la cardiotoxicité du traitement par l’oxaliplatine (risque d’allongement de l’intervalle QT [voir rubrique 4.4 du RMP de l’oxaliplatine]).
- Intervalle QT/QTc > 450 ms pour les hommes et > 470 ms pour les femmes.
- Hypertension non contrôlée (définie par une tension artérielle systolique > 140 mmHg et/ou une tension artérielle diastolique > 90 mmHg) ou antécédents de crise hypertensive (TA systolique > 20 mmHg) ou d’encéphalopathie hypertensive.
- Antécédents d’événements thromboemboliques veineux, y compris thrombose veineuse profonde et embolie pulmonaire, au cours du mois précédant l’inclusion dans l’étude.
- Antécédents d’accident vasculaire cérébral et/ou d’accident ischémique transitoire (AIT) au cours des 12 derniers mois.
- Toxicités persistantes liées à un traitement antérieur de grade supérieur à 1
- Persistance de symptômes cliniquement significatifs après un événement thromboembolique malgré un traitement anticoagulant.
- Thromboembolie artérielle (infarctus du myocarde, accident vasculaire cérébral, accident ischémique transitoire) survenant sous traitement antiangiogénique.
- Autres cancers actifs ou antécédents de cancer traités au cours des 5 dernières années, à l’exception du carcinome in situ du col de l’utérus ou du carcinome cutané basocellulaire ou squameux ou de tout autre carcinome in situ considéré comme guéri.
- Personnes privées de liberté ou sous tutelle ou incapables de donner leur consentement.
- Impossibilité de se soumettre au suivi médical de l’essai pour des raisons géographiques, sociales ou psychologiques.
Calendrier prévisionnel
Lancement de l’étude : Octobre 2025
Fin estimée des inclusions : Octobre 2027
Nombre de patients à inclure : 74
Coordonnateur de l'étude
Pr Jean-Marc PHELIP – CHU St Etienne
Promoteur de l'étude
Fédération Francophone de Cancérologie Digestive (FFCD)
Dernière mise à jour le 2 février 2026