IMMUNOLIFE2
Etude randomisée multicentrique de phase II évalunat la microbiothérapie fécale poolée par voie orale (MAAT033) concomittante au cemiplimab (CB) comparée au meilleur choix du médecin dans la résistance au blocage PD-1/PDL-1 en raison de la prise d’antibiotiques chez les patients avec un cancer bronchique non à petites cellules avancé
Type d'essai : Académique / Institutionnel
Etat de l'essai : Ouvert
Situation thérapeutique : Tumeur solide ( Métastatique / Rechute , Localement avancée / Non résécable )
Etablissement(s) participant(s)
Dr Stéphanie MARTINEZ
Détails de l'essai
Objectif principal
Evaluer si l’association de MaaT033 et de CB peut améliorer le taux de contrôle de la maladie (DCR) à 12 semaines par rapport au meilleur choix de l’investigateur (BIC) chez les patients réfractaires aux inhibiteurs du point de contrôle immunitaire (ICI) liés à la prise d’antibiotiques et atteints d’un CPNPC avancé. La confirmation de la réponse doit être démontrée par une évaluation 4 à 8 semaines après l’évaluation initiale de la réponse.
Objectif(s) secondaire(s)
Evaluer si l’association de MaaT033 et de CB peut avoir un impact sur le taux de réponse objective (ORR), la survie sans progression (PFS), la survie globale (OS), la durée de la réponse (DoR) et l’incidence des événements indésirables (EI) par rapport aux BIC.
Résumé / schéma de l'étude
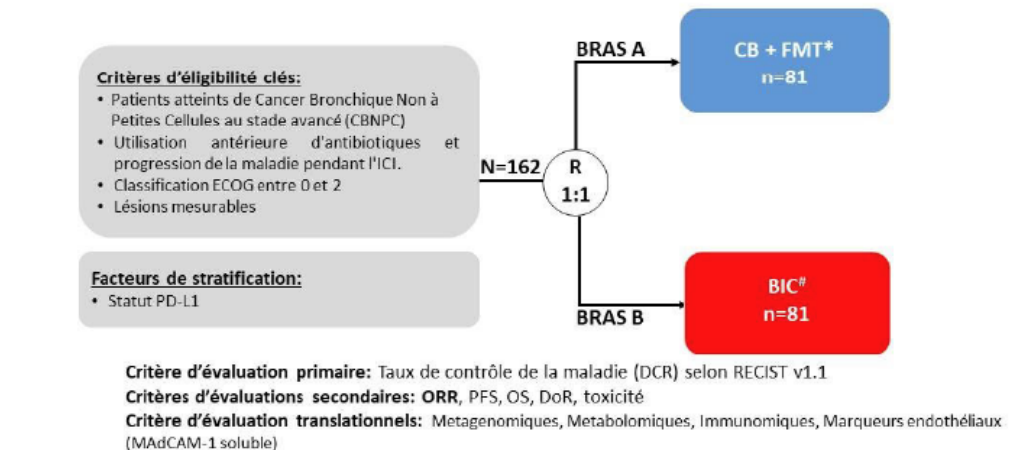
Critère(s) d'inclusion
- Participants who are at least 18 years of age on the day of signing informed consent.
- All participants must understand spoken and written national language.
- Histologically confirmed diagnosis of NSCLC (adenocarcinoma versus squamous cell carcinoma versus others
- Have metastatic or unresectable NSCLC and considered by their physician to be indicated for a new line of immunotherapy.
- Have an Eastern Cooperative Oncology Group performance status (ECOG-PS) of 0 to 2. Evaluation of ECOG-PS is to be performed within 7 days prior to the date of treatment allocation.
- Patients who have progressed after immunotherapy or immunotherapy plus platinum-based chemotherapy (with platinum-based chemotherapy and ICI either sequentially or concomitantly).
- Have received ATB within 60 days before and 42 days after the first ICI administration and have progressed within 6 months after the first ICI.
- There are no restrictions on the number of prior lines of treatment. Patients may be included regardless of the number of previous therapies received.
- A male participant must abstain from heterosexual activity or must agree to use a contraception as detailed below (or in Appendix 2 of this protocol) during the treatment period and for at least 9 months after the last dose of CB or BIC and refrain from donating sperm during this period. (In application of the new recommendations of the CTFG).
- A female participant is eligible to participate if she is not pregnant (see Appendix 2), not breastfeeding, and if at least one of the following conditions applies:
- Not a woman of childbearing potential (WOCBP) as defined in Appendix 2.
- A WOCBP should have a negative urine or serum pregnancy test within 72 hours prior to receiving the first dose of study medication. If the urine test is positive or cannot be confirmed as negative, a serum pregnancy test will be required. A WOCBP must agree to follow the contraceptive guidance in Appendix 2 or abstain from heterosexual activity during the treatment period and for at least 180 days, after the last dose of treatment.
- Patient should understand, sign, and date the written informed consent form prior to any protocol-specific procedures performed. Patient should be able and willing to comply with study visits and procedures as per protocol.
- Patients must be affiliated to a social security system or beneficiary of the sam
- Have an estimated life expectancy greater than 3 months (from inclusion).
- Meet acceptable steroid dose thresholds (i.e., not above the acceptable threshold <10 mg prednisone daily or equivalent) if receiving systemic steroids at physiologic dose
- Have measurable disease based on RECIST 1.1 criteria. Lesions situated in a previously irradiated area are considered measurable if progression has been demonstrated in such lesions.
- Have adequate organ function. All screening laboratory tests must be performed within 28 days prior to the start of study treatment.
Critère(s) de non-inclusion
- Immunodeficiency or systemic steroid therapy equivalent to prednisolone >10mg/day or equivalent within 7 days prior to the first dose of trial treatment.
- Active ongoing infection requiring ATB treatment.
- Has a known additional malignancy that is progressing or has required active treatment within the past 3 years prior to enrollment. Participants with basal cell carcinoma of the skin, squamous cell carcinoma of the skin, or carcinoma in situ (e.g. breast carcinoma, cervical cancer in situ) that have undergone potentially curative therapy are not excluded.
- Has received prior radiotherapy within 2 weeks of start of study treatment. Participants must have recovered from all radiation-related toxicities, not require corticosteroids, and not have had radiation pneumonitis. A 1-week washout is permitted for palliative radiation (≤2 weeks of radiotherapy) to non-central nervous system (CNS) disease.
- Has received a live vaccine within 30 days prior to the first dose of study drug. Examples of live vaccines include, but are not limited to, the following: measles, mumps, rubella, varicella/zoster (chicken pox), yellow fever, rabies, Bacillus Calmette-Guérin (BCG), and typhoid vaccine. Seasonal influenza vaccines for injection are generally killed virus vaccines and are allowed; however, intranasal influenza vaccines (eg, FluMist®) are live attenuated vaccines and are not allowed.
- Is currently participating in or has participated in a study of an investigational agent or has used an investigational device within 4 weeks prior to the first dose of study treatment. Participants who have entered the follow-up phase of an investigational study may participate if it has been 4 weeks after the last dose of the previous investigational agent and that all study drug-related AEs have resolved to grade 1 or less.
- Has known active CNS metastases and/or carcinomatous meningitis. Participants with previously treated brain metastases may participate provided they are radiologically stable, i.e. without evidence of progression for at least 4 weeks by repeat imaging (note that the repeat imaging should be performed during study screening), clinically stable and without requirement of steroid treatment for at least 14 days prior to first dose of study treatment.
- Has active autoimmune disease that has required systemic treatment in the past 2 years (i.e. with use of disease modifying agents, corticosteroids or immunosuppressive drugs). Replacement therapy (e.g., thyroxine, insulin, or physiologic corticosteroid replacement therapy for adrenal or pituitary insufficiency, etc.) is not considered a form of systemic treatment.
- Has a known history of Human Immunodeficiency Virus (HIV).
- Has a known history of Hepatitis B virus (HBV, defined as Hepatitis B surface antigen [HBsAg] reactive) or known active Hepatitis C virus (HCV, considered active if HCV RNA is detected) infection. Note: no testing for HBV and HCV is required unless mandated by local health authority.
- Has a history or current evidence of any condition, therapy, or laboratory abnormality that might confound the results of the study, interfere with the subject’s participation for the full duration of the study, or is not in the best interest of the subject to participate, in the opinion of the treating investigator.
- Any condition which, in the Investigator’s opinion, makes it undesirable for the subject to participate in a clinical trial or which would jeopardize compliance with the protocol.
- Patient under guardianship or deprived of his liberty by a judicial or administrative decision or incapable of giving its consent
- Is pregnant or breastfeeding or expecting to conceive or father children within the projected duration of the study, starting with the screening visit through 180 days after the last dose of treatment.
- Persistent toxicities related to prior treatment of grade greater than 1.
- Swallowing disorders which can affect the intake of the oral pooled fecal microbiotherapy (MaaT033).
Calendrier prévisionnel
Lancement de l’étude : Septembre 2025
Fin estimée des inclusions : Septembre 2027
Nombre de patients à inclure : 162
Coordonnateur de l'étude
Dr Lisa De Rosa – Gustave Roussy – CLCC Villejuif
Promoteur de l'étude
Gustave Roussy- CLCC Villejuif