PRODIGE 98 -FFCD 2105 - AMPIRINOX
Etude de phase III randomisée multicentrique comparant une chimiothérapie adjuvante de 6 mois par FOLFIRINOX modifié versus une monochimiothérapie par capécitabine ou gemcitabine chez les patients opérés d’un adénocarcinome de l’ampoule de Vater
Phase : III
Type d'essai : Académique / Institutionnel
Etat de l'essai : Ouvert
Situation thérapeutique : Tumeur solide ( Adjuvant )
Type d'essai : Académique / Institutionnel
Etat de l'essai : Ouvert
Situation thérapeutique : Tumeur solide ( Adjuvant )
Etablissement(s) participant(s)
Pr Laetitia DAHAN
Dr Julie SIGRAND
Détails de l'essai
Objectif principal
To assess efficacy of adjuvant mFOLFIRINOX versus single-agent chemotherapy (gemcitabine or capecitabine) in improving disease-free survival (DFS) after surgical resection of an ampullary adenocarcinoma.
Objectif(s) secondaire(s)
- Overall survival (OS).
- Rate of patients completing 3 and 6-month chemotherapy schedule according to percentage of administered dose of each product.
- Assessment of quality of life by EORTC QLQ-C30 and QLQ-PAN26.
- Assessment of toxicities according to NCI-CTCAE v5.0.
- Subgoup analyses on OS and DFS by prespecified subgroups.
Résumé / schéma de l'étude
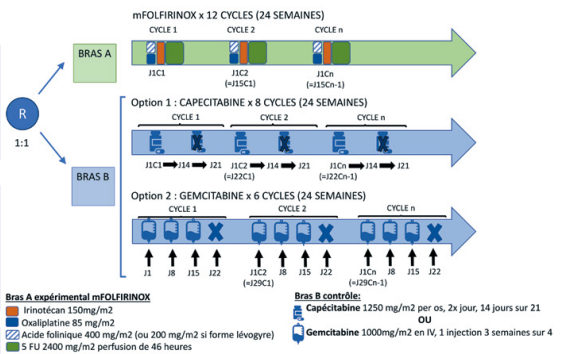
Critère(s) d'inclusion
- Histologically proven adenocarcinoma on surgical specimen.
- Macroscopically complete surgical resection of an ampullary adenocarcinoma (R0 or R1).
- Adenocarcinoma removed within 12 weeks prior to enrollment.
- Patients ≥ 18 years of age.
- Patient without metastatic disease on CT scan < 4 weeks prior to inclusion.
- WHO performance status 0 or 1 (WHO 0 if age >75).
- Normal values of kalemia, magnesemia and calcemia.
- Patient able to understand and sign the information and informed consent note.
- Women of childbearing age and men who are sexually active with women of childbearing age must agree to use highly effective contraception during the trial treatment at least until 6 months after the end of the experimental treatment. Women of childbearing potential must use highly effective contraception at least until 9 months after the end of the treatment with oxaliplatin.
- Patient affiliated to a social security scheme for France, or equivalents in European countries.
- CA19.9 level < 180 U/L at inclusion (post-operative level).
Critère(s) de non-inclusion
- Neoadjuvant systemic chemotherapy.
- pT1N0M0 tumors.
- Active infection by HBV, HCV or HIV.
- Dihydropyrimidine dehydrogenase deficiency (uracilemia ≥ 16 ng/mL).
- Pre-existing peripheral neuropathy (grade ≥ 2).
- Unresolved or uncontrolled concomitant medical conditions.
- Neutrophils < 1500/mm3, platelets < 150 000/mm3, Haemoglobin < 9 g/dL.
- Total bilirubin > 1.5 x normal.
- Creatinine clearance < 50 ml/min according to MDRD.
- AST or ALT > 2.5 x UNL, alkaline phosphatase > 2.5 x normal at least 15 days after resection.
- Patients with poor nutritional status represented by albuminemia < 30.0g/dl.
- History of myocardial infarction within the last 6 months, severe coronary artery disease or severe heart failure.
- Active and/or potentially severe infection.
- Treatment with a strong cytochrome P450 inhibitor within 4 weeks prior to the administration of the protocol treatment (Treatment with Hypericum perforatum).
- Patient under treatment by brivudine, or treated by brivudine within 4 weeks prior to beginning of study treatment.
- Concomitant use with St John’s Wort.
- QT/QTc interval longer than 450msec for men and longer than 470msec for women on the ECG.
- Hypersensitivity to any of the study products or their excipients.
- Administration of live vaccines within 28 days prior to randomization.
- Other cancer treated within the last 5 years except adequately treated, in situ cervical carcinoma or basocellular/spinocellular carcinoma.
- Chronic bowel disease requiring specific treatment and/or intestinal obstruction.
- Pregnant or breastfeeding woman.
- Person under guardianship.
- Inability to undergo the medical follow-up of the trial for geographical, social or psychological reasons.
Calendrier prévisionnel
Lancement de l’étude : Mai 2025
Fin estimée des inclusions : Mai 2029
Nombre de patients à inclure : 292
Coordonnateur de l'étude
Dr Gaël ROTH – CHU de Grenoble
Promoteur de l'étude
Fédération Francophone de Cancérologie Digestive (FFCD)
Dernière mise à jour le 24 septembre 2025