ICARUS-BREAST01
Etude de phase II, ouverte, visant à évaluer l’efficacité du U3-1402, un anticorps conjugue anti-HER3, chez les patients atteints d’un cancer du sein avancé avec analyses des biomarqueurs associés à la réponse au traitement
Type d'essai : Académique / Institutionnel
Etat de l'essai : Ouvert
Situation thérapeutique : Tumeur solide ( Métastatique / Rechute , Localement avancée / Non résécable )
Etablissement(s) participant(s)
Dr Agnès DUCOULOMBIER
Dr Cécile VICIER
Détails de l'essai
Objectif principal
L’objectif de l’étude est d’évaluer l’efficacité du U3-1402, un anticorps conjugué anti-HER3, chez des patients ayant un cancer du sein avancé.
Objectif(s) secondaire(s)
Evaluer l’efficacité de l’U3-1402 en termes de taux de réponse objective en fonction du niveau d’expression du récepteur HER3, durée de réponse, survie sans progression, taux de bénéfice clinique et survie globale.
Evaluer la sécurité et la tolérabilité de l’U3-1402 en termes d’événements indésirables, anomalies biologiques (critères NCI-CTCAE v5.0), anomalies dans les électrocardiogrammes et changements de statut de performance ECOG.
Evaluer les résultats rapportés par les patients : score du fonctionnement physique de l’Organisation Européenne pour la Recherche et le Traitement du Cancer (EORTC) Questionnaire de qualité de vie (QLQ-C30) ; Etat de santé global/échelle de la qualité de vie (EORTC QLQ-C30), échelle des symptômes du cancer du sein (EORTC QLQ-BR23).
Décrire la dynamique de l’expression de HER3 après une ou plusieurs doses d’U3-1402 (exploratoire).
Identifier les prédicteurs moléculaires de la survie sans progression et de la survie globale (exploratoire).
Décrire les effets immunitaires (par exemple : mort cellulaire immunogène, etc.), l’effet bystander et le stress de réplication induits par l’U3-1402 (exploratoire).
Décrire les mécanismes moléculaires de la résistance primaire et secondaire (exploratoire).
Evaluer les niveaux de cellules tumorales circulantes pendant le traitement avec U3-1402 (exploratoire).
Evaluer la pharmacocinétique de l’U3-1402 et les anticorps anti-médicament et les relations dose-effet de l’U3-1402 (exploratoire).
Résumé / schéma de l'étude
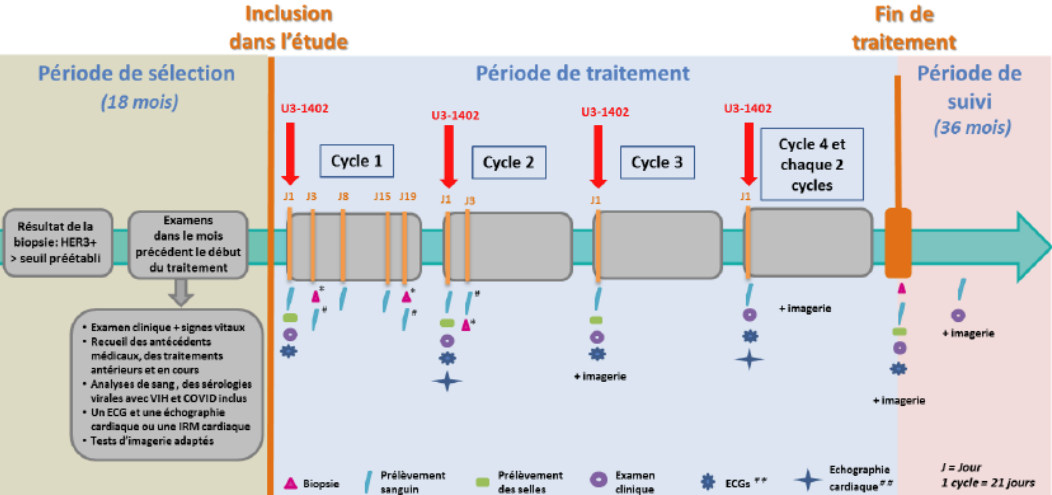
Critère(s) d'inclusion
- Patient de sexe féminin ou masculin âgés de 18 ans ou plus.
- Patient avec cancer du sein confirmé histologiquement non résécable localement avancé ou métastatique avec au moment du diagnostic, une surexpression de HER3 (positivité de la membrane sur ≥75% des cellules tumorales à 10x objectif) sur le plus récent échantillon de tissu tumoral disponible. Les tumeurs doivent également être positives pour les récepteurs hormonaux (récepteurs des œstrogènes positifs (RE+) et/ou récepteurs de la progestérone (RP+)) et HER2- negative (IHC2+ / ISH négatif ou IHC1 + ou IHC0 +).
Si la tumeur était ER+ et/ou PR+ au moment du diagnostic et est devenue ER- et/ou PR- dans les biopsies suivantes, le patient peut être inclut dans l’étude. - Patient avec une progression radiologique documentée non résécable ou métastatique.
- Le patient peut avoir reçu des anthracyclines et des taxanes comme traitement (néo) adjuvant (qui ne compte pas comme une ligne de traitement) et doit avoir reçu 1 ligne de chimiothérapie pour cancer du sein avancé, mais pas plus d’une ligne. Le patient doit avoir une progression de la tumeur confirmée cliniquement ou radiologiquement suite aux traitements inhibiteurs de CDK4/6 combinés aux traitements endocriniens. Ces traitements ne doivent pas nécessairement être la dernière ligne de traitement avant la participation du patient à l’étude. Les traitements antérieurs avec des inhibiteurs de phosphoinositide 3-kinase (PI3K), des inhibiteurs de cible de la rapamycine chez les mammifères (mTOR), des inhibiteurs de l’AKT et des inhibiteurs de la Poly (ADP-ribose) polymérase (PARP) sont autorisés.
- Le patient doit avoir un site métastatique facilement accessible pour la biopsie (à l’exception des métastases osseuses) et doit avoir accepté d’effectuer des biopsies avant et pendant le traitement.
- Le patient doit avoir au moins une lésion mesurable radiologiquement (différente du site de biopsie) selon les critères RECIST V1.1.
- Le patient doit avoir un ECOG PS 0 ou 1 au moment du dépistage.
- Le patient doit avoir une espérance de vie ≥ 12 semaines.
- Possède une réserve de moelle osseuse et une fonction organique adéquates, sur la base des données de laboratoire locales dans les 14 jours précédant le cycle 1, jour 1, défini comme :
- Numération plaquettaire ≥100 000/mm3 ou ≥100 × 109/L (les transfusions de plaquettes ne sont pas autorisées jusqu’à 14 jours avant le cycle 1 jour 1 pour répondre à l’admissibilité).
- Hémoglobine :≥ 9,0 g / dL (la transfusion et/ou le soutien des facteurs de croissance sont autorisés).
- Neutrophiles : ≥ 1500 / mm3 ou ≥ 1,5 × 109/L (l’utilisation de facteurs de croissance n’est pas autorisée dans les 15 jours précédant le cycle 1).
- Créatine sérique : CrS ≤ 1,5 × LSN, OU clairance de la créatinine (ClCr) ≥ 30 mL/min, calculée à l’aide de l’équation de Cockcroft-Gault ou de la ClCr mesurée ; la confirmation de ClCr n’est requise que lorsque CrS est> 1,5 × LSN.
- ASAT / ALAT : ≤ 3 × LSN (si des métastases hépatiques sont présentes, ≤5 × LSN).
- Bilirubine totale : ≤ 1,5 × LSN en l’absence de métastases hépatiques (<3 × LSN en présence d’un syndrome de Gilbert documenté (hyperbilirubinémie non conjuguée) ou de métastases hépatiques.
- Albumine sérique : ≥ 2.5 g/dL.
- Potassium : ≥ LIN du laboratoire local.
- Magnésium : ≥ LIN du laboratoire local.
- Calcium : Calcémie corrigée ≥ LIN selon un laboratoire local.
- Temps de prothrombine (TP) ou temps de prothrombine – rapport international normalisé (TP-RIN) et temps de thromboplastine partielle activée (aTTP) / temps de thromboplastine partielle (TTP) : ≤ 1,5 × (LSN), sauf pour les sujets sous anticoagulants dérivés de la coumarine ou autre traitement anticoagulant similaire, qui doivent avoir un TP-RIN dans la plage thérapeutique jugée appropriée par l’investigateur.
- Les patientes en âge de procréer doivent avoir un test de grossesse négatif lors du dépistage, soit sérique 14 jours précédents la première administration du traitement ou urinaire 72 heures avant la première administration du traitement. Les patientes en âge de procréer doivent accepter d’utiliser une forme de contraception hautement efficace ou d’éviter les rapports sexuels pendantet à la fin de l’étude et pendant au moins 7 mois après la dernière dose du médicament à l’étude.
- S’il s’agit d’un homme, le patient doit être chirurgicalement stérile, il doit s’abstenir de tout rapport hétérosexuel ou il doit être disposé à utiliser une méthode contraceptive hautement efficace lors de son inscription, pendant la période de traitement et pendant au moins 4 mois après la dernière dose du médicament à l’étude. Les patients masculins ne doivent pas congeler ni donner de sperme à partir du dépistage et pendant toute la durée de l’étude, et pendant au moins 4 mois après l’administration finale du médicament à l’étude.
- Le patient doit comprendre, signer et dater le consentement éclairé écrit avant d‘effectuer toute procédure spécifique au protocole. Le patient doit être capable et disposé à se conformer aux visites d’étude et à la procédure conformément au protocole.
- Affilié à un système de sécurité sociale ou bénéficiaire du même.
Critère(s) de non-inclusion
- Patiente avec un cancer du sein susceptible de résection ou de radiothérapie à visée curative.
- Patient ayant des antécédents de pneumopathie interstitielle (PI) (y compris fibrose pulmonaire ou pneumopathie par irradiation), ou une PI actuelle ou est suspecté de présenter une PI par imagerie pendant le dépistage.
- Patient présentant une atteinte pulmonaire cliniquement sévère (sur la base de l’évaluation de l’investigateur) résultant de maladies pulmonaires intercurrentes, y compris, mais sans s’y limiter :
- Toute maladie pulmonaire sous-jacente (par exemple, embolie pulmonaire, asthme sévère, maladie pulmonaire obstructive chronique sévère (MPOC), maladie pulmonaire restrictive, épanchement pleural);
- Toutes maladies auto-immunes du tissu conjonctif ou inflammatoire avec atteinte pulmonaire (par exemple, polyarthrite rhumatoïde, syndrome de Sjögren, sarcoïdose) OU une pneumonectomie antérieure.
- Patient recevant des doses de corticostéroïdes systémiques chroniques de >10 mg de prednisone ou équivalent, ou toute forme de traitement immunosuppresseur avant le cycle 1 jour 1. Les sujets nécessitant l’utilisation de bronchodilatateurs, de stéroïdes inhalés ou d’injections locales de stéroïdes peuvent être inclus dans l’étude.
- Patient présentant des signes de maladie leptoméningée.
- Patient atteint d’une maladie cornéenne cliniquement significative.
- Tout signe de maladies systémiques graves ou incontrôlées (y compris les diathèses hémorragiques actives, une infection active, ou une maladie psychiatrique) qui, de l’avis de l’investigateur, ne souhaitent pas que le sujet participe à l’étude ou qui compromettrait le respect du protocole). Le dépistage des maladies chroniques n’est pas nécessaire.
- Patient présentant une compression de la moelle épinière ou des métastases cérébrales cliniquement actives, défini comme non traitées et symptomatiques, ou ayant besoin d’un traitement par corticostéroïdes ou anticonvulsivants pour contrôler les symptômes associés. Les patients présentant des métastases cérébrales cliniquement inactives ou traitées qui sont asymptomatiques (c’est-à-dire, sans signes ou symptômes neurologiques et n’ayant pas besoin d’un traitement par corticostéroïdes ou anticonvulsivants) peuvent être inclus dans l’étude. Les patients doivent avoir un état neurologique stable pendant au moins 2 semaines avant le cycle 1 jour 1.
- Période de lavage inadéquate avant le cycle 1 jour 1, définie comme :
- Une radiothérapie au cerveau en entier <14 jours ou radiothérapie stéréotaxique cérébral < 7 jours.
- Toute chimiothérapie cytotoxique, agents expérimentaux ou autre(s) médicament(s) anticancéreux issu d’un schéma de traitement anticancéreux ou d’une étude clinique antérieure (autre que l’inhibiteur tyrosine kinase du récepteur du facteur de croissance épidermique (ITK EGFR), < 14 jours ou 5 demi-vies, selon la plus longue des deux.
- Traitement avec immunothérapie < 21 jours.
- Traitement avec hormonothérapie < 21 jours.
- Chirurgie majeure (à l’exclusion de la mise en place de l’accès vasculaire) <28 jours
- Traitement de radiothérapie à plus de 30% de la moelle osseuse ou avec un large champ de rayonnement < 28 jours ou radiothérapie palliative < 14 jours.
- Chloroquine ou hydroxychloroquine ≤ 14 jours.
- Administration d‘un vaccin vivant < 28 jours.
- Traitement antérieur avec un anticorps anti-HER3 et/ou un anticorps conjugué (AC) qui consiste en un dérivé d’exatécan qui est un inhibiteur de la topoisomérase I (par exemple, le trastuzumab deruxtecan).
- La présence des toxicités (autres que l’alopécie) non résolues, grade ≤2 selon les critères de terminologie communs du NCI-CTCAE version 5.0, et résultant d’un traitement anticancéreux précédent.
- La présence d’une hypersensibilité aux substances médicamenteuses ou aux ingrédients inactifs du produit pharmaceutique.
- La présence d’une hypersensibilité à d’autres anticorps monoclonaux.
- La présence d’une malignité primaire autre que le cancer du sein, localement avancé ou métastatique dans les 3 ans précédant du cycle 1 jour 1, à l’exception d’un cancer de la peau non mélanique réséqué de manière adéquate, d’un cancer in situ traitée de manière curative ou d’autres tumeurs solides traitées de manière curative.
- Maladie cardiovasculaire non contrôlée ou significative avant le premier jour du cycle 1, y compris :
- Intervalle QT corrigé selon la formule de Fridericia (QTcF) > 470 ms chez les femmes et > 450 ms chez les hommes (évalué en fonction de trois ECGs, à environ une minute d’intervalle).
- FEVG <50% par ECHO ou par IRM cardiaque si indiqué par l’investigateur ou le cardiologue soignant.
- Hypertension non contrôlée (pression artérielle systolique au repos >140 mmHg ou pression artérielle diastolique >90 mmHg).
- Infarctus du myocarde dans les 6 mois précédents.
- Insuffisance cardiaque congestive symptomatique, Classe II à IV sur l’échelle de NYHA, dans les 28 jours précédents.
- Angine de poitrine incontrôlée dans les 6 mois précédents.
- Arythmie cardiaque nécessitant un traitement antiarythmique.
- Hépatite B et / ou hépatite C active(s), présentant des signes sérologiques d’infection virale dans les 28 jours avant le cycle 1, jour 1
Les patients ayant des antécédents d’infection par le virus de l’hépatite B (VHB) résolue sont éligibles si :- Antigène de surface de l’hépatite (HBsAg) négatif et anticorps de l’hépatite B (anti-HBc) positifs;
OU - La charge virale HBsAg est positive et HBV DNA est documentée comme étant ≤ 2000 UI/mL en l’absence de traitement antiviral et au cours des 12 semaines précédant l’évaluation de la charge virale avec des transaminases normales (en l’absence de métastases hépatiques) ; OU c. La charge virale HBsAg est positive et HBV DNA est documentée comme étant ≤ 2000 UI/mL en l’absence de traitement antiviral et au cours des 12 semaines précédant l’évaluation de la charge virale avec métastases hépatiques et transaminases anormales AST / ALT < 3 LSN.
Les patients ayant des antécédents d’infection par l’hépatite C ne seront éligibles à l’enrôlement que si la charge virale, selon les normes locales de détection, est documentée comme étant inférieure au niveau de détection en l’absence de traitement antiviral au cours des 12 semaines précédentes (par exemple : réponse virale soutenue selon l’étiquette du produit local mais pas moins de 12 semaines, selon la plus longue des deux).
- Antigène de surface de l’hépatite (HBsAg) négatif et anticorps de l’hépatite B (anti-HBc) positifs;
- Femme enceinte ou qui allaite ou qui a l’intention de devenir enceinte pendant l’étude.
- Patients atteints du virus de l’immunodéficience humaine (VIH) ou du COVID-19.
- Patient présentant une affection psychologique, familiale, sociologique ou géographique susceptible d’entraver le respect des procédures du protocole de l’étude et du calendrier de suivi.
- Patient sous tutelle ou privé de sa liberté par décision judiciaire ou administrative ou incapable de donner son consentement.
- Participation à une autre étude clinique évaluant un médicament expérimental (sauf recherche non interventionnelle).
Calendrier prévisionnel
Lancement de l’étude : Mai 2021
Fin estimée des inclusions : Avril 2027
Nombre de patients à inclure : 139
Coordonnateur de l'étude
Dr Barbara PISTILLI – Gustave Roussy – CLCC Villejuif
Promoteur de l'étude
Gustave Roussy – CLCC Villejuif