PRODIGE 107 – FFCD 2306 – COLOSOTO
Etude de phase II monobras évaluant le 5-fluorouracile associé au panitumumab (anti-EGFR) et au Sotorasib (inhibiteur de KRAS G12C) dans le traitement de première intention des patients non éligibles à une chimiothérapie double/triplet présentant un adénocarcinome colorectal avancé et non résécable avec mutation de KRAS G12C
Type d'essai : Académique / Institutionnel
Etat de l'essai : Ouvert
Situation thérapeutique : Tumeur solide ( Métastatique / Rechute , Localement avancée / Non résécable )
Cibles / marqueurs : KRAS G12C
Etablissement(s) participant(s)
Dr Nicolas BARRIERE
Pr Laetitia DAHAN
Détails de l'essai
Objectif principal
Survie sans progression sous 5-fluorouracile plus Panitumumab et Sotorasib (inhibiteur de KRAS G12C) à 8 mois dans le traitement de première ligne des patients non éligibles pour une double/triple chimiothérapie avec un adénocarcinome colorectal avancé non résécable muté KRAS G12C (basé sur RECIST 1.1 évalué par l’investigateur).
Objectif(s) secondaire(s)
Mediane de survie sans progression.
Taux de contrôle de la maladie.
Temps jusqu’à progression.
Survie Gloable.
Taux de meilleure réponse.
Durée de la réponse.
Toxicité.
Questionnaires de qualité de vie et FACIT-GP5.
Evaluation gériatrique (G8 et G-CODE).
Résumé / schéma de l'étude
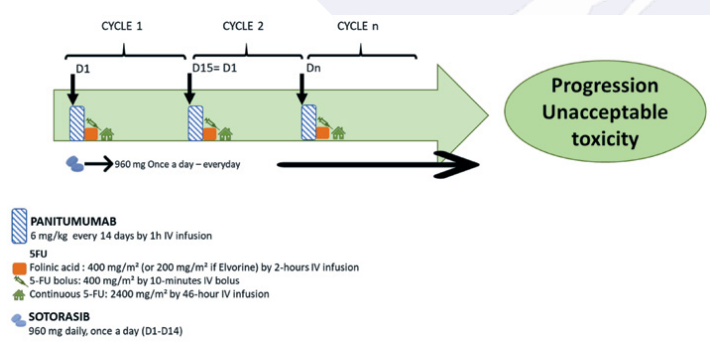
Critère(s) d'inclusion
- Age ≥ 18 ans.
- Adénocarcinome colorectal non résécable, localement avancé ou métastatique, histologiquement prouvé à un stade avancé.
- Mutation G12C du KRAS prouvée, évaluée localement au moyen d’un test conforme à l’IVDR.
- Accord du patient pour participer aux études biologiques (échantillons de sang pour analyse ADNtc et échantillon de tumeur).
- Patient non éligible au traitement double/triplet :
a. Patients avec un PS=2 de l’OMS
b. Patients âgés de 70 à 75 ans avec un PS = 1 de l’OMS
c. Patients ≥ 75 ans - Lésion mesurable selon les critères RECIST 1.1
- Pas de traitement antérieur de la maladie métastatique. Une chimiothérapie adjuvante antérieure est autorisée s’il s’est écoulé plus de 6 mois entre la fin du traitement adjuvant et la rechute.
- Fonction des organes adéquate: Hémoglobine > 9 g/dl, neutrophiles > 1500 /mm3, plaquettes > 80 000/mm3, taux de clairance de la créatinine ≥50 ml/min calculé selon la formule MDRD, ALT/AST ≤ 5×ULN et bilirubine totale ≤ 1,5×ULN.
- Capacité à comprendre et à signer un consentement pour participer à l’étude.
- Fournir un consentement éclairé pour la participation à l’étude.
- Espérance de vie > 6 mois.
- Les femmes en âge de procréer doivent accepter d’utiliser un moyen de contraception pendant l’essai et pendant au moins 6 mois après l’arrêt des traitements expérimentaux.
Les hommes qui ont des relations sexuelles avec des femmes en âge de procréer doivent accepter d’utiliser un moyen de contraception pendant le traitement et pendant au moins 3 mois après l’arrêt des traitements expérimentaux. - Patient affilié à un régime de sécurité sociale.
Critère(s) de non-inclusion
- Patient répondant l’un des critères suivants :
- Patient apte à recevoir un régime doublet/triplet.
- Patient avec un PS 3 ou 4 de l’OMS.
- Patient < 75 ans avec un PS = 0 de l’OMS.
- Patient < 70 ans avec un PS de 0 ou 1 de l’OMS.
- Maladie intercurrente non contrôlée incluant une insuffisance hépatique (cirrhose du foie Child Pugh B ou C) et pulmonaire (volume expiratoire forcé d’une seconde < 50%) sévère.
- Les patients dont la tumeur présente une instabilité des microsatellites (MSI-H) ou un statut de réparation des mésappariements déficient (dMMR).
- Anomalies cardiaques cliniquement significatives, y compris antécédents de l’un des éléments suivants : cardiomyopathie sévère, insuffisance cardiaque congestive de grade ≥3 de la New York Heart Association, antécédents de maladie cardiovasculaire athéroscléreuse cliniquement significative (c’est-à-dire active) (infarctus du myocarde, angor instable, accident vasculaire cérébral dans les 6 mois précédant la première dose des traitements à l’étude).
- Patients présentant un déficit enzymatique en dihydropyrimidine déshydrogénase (DPD) (uracilémie ≥ 16 ng/mL).
- Immunothérapie dans les 3 mois précédant le début de l’étude de traitement.
- Patient sous traitement par des inducteurs puissants du CYP3A4.
- Patients traités à la brivudine dans les 4 semaines précédant la première dose du traitement à l’étude, ou traitement concomitant à la brivudine.
- Patient souffrant d’une infection potentiellement grave.
- Administration d’un vaccin vivant ou vivant atténué dans les 30 jours précédant le début de la première dose du traitement à l’étude.
- Mauvais état nutritionnel (albuminémie < 25 g/l ou perte de poids > 10 % au cours du dernier mois).
- Problèmes héréditaires d’intolérance au galactose, de déficit en lactase totale ou de malabsorption du glucose et du galactose.
- Autre tumeur maligne dans les 2 ans précédant l’inscription à l’étude, à l’exception d’un cancer localisé in situ, d’un cancer de la peau basocellulaire ou squameux traité de manière adéquate.
- Moins de 4 semaines après une intervention chirurgicale majeure et pas suffisamment rétabli de l’intervention et/ou de toute complication liée à l’intervention.
- Patients présentant des toxicités persistantes liées à un traitement antérieur de grade supérieur à 1.
- Participer ou avoir participé à une étude portant sur un agent expérimental ou avoir utilisé un dispositif expérimental dans les 3 semaines ou 5 demi-vies (selon la durée la plus longue) précédant l’entrée dans l’étude.
- Hypersensibilité à l’une des substances actives ou à l’un des excipients des traitements de l’essai.
- Patients ayant des antécédents de pneumopathie interstitielle ou de fibrose pulmonaire.
- Patient souffrant d’une maladie pulmonaire interstitielle ou d’une fibrose pulmonaire.
- Patient souffrant d’un trouble psychiatrique ou d’un abus de substances connu qui pourrait interférer avec la capacité du patient à coopérer avec les exigences de l’étude.
- Patient sous protection judiciaire et patient légalement institutionnalisé ou sous tutelle ou incapable de donner son consentement.
- Femme enceinte ou allaitante.
- Incapacité de se soumettre au suivi médical de l’essai pour des raisons géographiques, sociales ou psychologiques.
Calendrier prévisionnel
Lancement de l’étude : Septembre 2025
Fin estimée des inclusions : Septembre 2028
Nombre de patients à inclure : 37
Coordonnateur de l'étude
Pr David TOUGERON – CHU de Poitiers
Promoteur de l'étude
Fédération Francophone de Cancérologie Digestive (FFCD)