IFCT-2402 LAG-MAPS
Essai de phase IIR non-comparatif évaluant le fianlimab en association avec le cemiplimab et une chimiothérapie platine-pemetrexed ou le cemiplimab et une chimiothérapie platine-pemetrexed chez des patients naïfs de traitement présentant un mésothéliome pleural
Type d'essai : Académique / Institutionnel
Etat de l'essai : Ouvert
Situation thérapeutique : Tumeur solide ( Métastatique / Rechute , Localement avancée / Non résécable )
Etablissement(s) participant(s)
Dr Marie BERNARDI
Dr Jacques LE TREUT
Pr Laurent GREILLIER
Dr Clarisse AUDIGIER-VALETTE
Pr Olivier BYLICKI
Dr Nicolas CLOAREC
Détails de l'essai
Objectif principal
Activité de la combinaison double immunothérapie anti-LAG3/anti-PD-1 et chimiothérapie platine/pemetrexed mesurée par le taux de contrôle de la maladie (DCR) à 6 mois, évaluée par une revue centralisée.
Objectif(s) secondaire(s)
Tolérance et sécurité du traitement dans les deux bras de l’étude.
Activité de la combinaison double immunothérapie anti-LAG3/anti-PD-1 et chimiothérapie platine/pemetrexed mesurée par :
– La survie sans progression (PFS) évaluée par l’investigateur et par une revue centralisée.
– La PFS en fonction du sous-type histologique.
– La survie globale.
– Le taux de réponse global.
Qualité de vie.
Résumé / schéma de l'étude
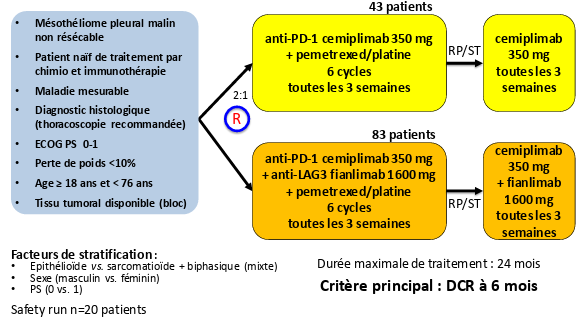
Critère(s) d'inclusion
- Consentement éclairé signé :
- Les patients doivent avoir signé et daté un formulaire de consentement éclairé écrit approuvé par un comité d’éthique conformément aux directives réglementaires. Ce consentement doit être obtenu avant la réalisation de toute procédure liée au protocole qui ne fait pas partie de la prise en charge standard du patient.
- Les patients doivent être désireux et capables de se conformer aux visites prévues, au planning de traitement et aux tests de laboratoire.
- Diagnostic histologique (pas de cytologie autorisée, biopsie par thoracoscopie recommandée).
- Mésothéliome pleural non résécable, évalué par une Réunion de Concertation Pluridisciplinaire (RCP) spécialisée d’oncologie thoracique comprenant un chirurgien thoracique spécialisé.
- Maladie mesurable par tomodensitométrie avec injection d’iode selon les critères RECIST 1.1 modifiés pour le mésothéliome (épaisseur pleurale perpendiculaire à la paroi thoracique ou au médiastin de 7 mm ou plus, sur 2 positions, à 3 niveaux distincts sur des coupes transversales de tomodensitométrie, séparées d’au moins 1 cm, la somme de 6 mesures définissant une mesure pleurale unidimensionnelle), ou selon les critères RECIST 1.1 pour les ganglions médiastinaux ou les lésions métastatiques.
- ECOG PS 0 et 1.
- Perte de poids <10 % dans les 3 mois précédant l’entrée dans l’étude.
- Patient naïf de traitement (chimiothérapie et immunothérapie).
- Age ≥ 18 ans, < 76 ans.
- Espérance de vie > 3 mois.
- Échantillons tumoraux disponibles (au moins 10 lames de l’échantillon de biopsie pleurale par thoracoscopie).
- Fonctions biologiques adéquates : clairance de la créatinine ≥45 ml/min (Cockroft ou MDRD ou CKD-epi) ; neutrophiles ≥ 1500/mm3 ; plaquettes ≥ 100000/mm3 ; hémoglobine ≥ 9g/dL ; ASAT et ALAT < 3 x ULN, bilirubine totale < 2 x ULN (les patients présentant des métastases hépatiques ou un syndrome de Gilbert doivent avoir des ASAT et ALAT ≤ 5 x ULN et une bilirubine totale de base ≤ 2 x ULN).
- Les femmes qui sont susceptibles de procréer (physiologiquement apte à mener une grossesse) doivent avoir un sérum (bêta-hCG) négatif lors du dépistage. Les femmes susceptibles de procréer sont définies comme des femmes fertiles depuis les premières règles jusqu’à la ménopause, à moins qu’elles ne soient définitivement stériles. Les méthodes de stérilisation permanente comprennent : l’hystérectomie, la salpingectomie bilatérale et l’ovariectomie bilatérale. L’état post-ménopausique est défini par l’absence de menstruations pendant 12 mois sans cause médicale. Un taux élevé de FSH post-ménopause peut confirmer un état post-ménopausique chez les femmes qui n’utilisent pas de contraception hormonale ou de traitement hormonal de substitution. Cependant, en l’absence de 12 mois d’aménorrhée, une seule mesure de la FSH est insuffisante pour déterminer l’apparition d’un état post-ménopausique. Les définitions ci-dessus sont conformes aux recommandations du CTFG. Les tests de grossesse et la contraception ne sont pas nécessaires pour les femmes ayant subi une hystérectomie ou une ligature des trompes documentée.
- Les participants masculins à l’étude ayant des partenaires susceptibles de procréer doivent utiliser des préservatifs pendant l’étude et jusqu’à 6 mois après la dernière dose du traitement de l’étude, à moins qu’ils ne soient vasectomisés ou qu’ils pratiquent l’abstinence sexuelle.
- Le partenaire vasectomisé ou le patient vasectomisé doit avoir fait l’objet d’une évaluation médicale de la réussite de l’intervention chirurgicale. NB : L’abstinence périodique (méthodes calendaires, sympto-thermiques, postovulation), le retrait (coït interrompu), les spermicides uniquement et la méthode de l’aménorrhée lactationnelle ne sont pas des méthodes de contraception acceptables. Le préservatif féminin et le préservatif masculin ne doivent pas être utilisés ensemble.
- Les femmes susceptibles de procréer doivent s’engager à ne pas donner d’ovules (ovules, ovocytes) à des fins de procréation assistée pendant toute la durée de l’essai et jusqu’à 6 mois après le dernier traitement.
Les hommes doivent accepter de ne pas donner de sperme pendant l’essai et pendant les 6 mois suivant la dernière dose de traitement. - Comme le recommandent les référentiels français actuels, une radiothérapie pour les voies de thoracocentèse (3 x 7Gy) est réalisée pour les patients ayant subi une thoracocentèse ou une thoracoscopie dans les deux mois précédant l’inclusion, avec un intervalle fermement recommandé entre la procédure de thoracoscopie (l’ablation des drains) et l’irradiation ne dépassant pas 42 jours. Un intervalle de 7 jours entre la fin de la radiothérapie et le début du traitement doit être respecté.
Critère(s) de non-inclusion
- ECOG PS>2.
- Traitement antérieur du cancer, y compris chimiothérapie ou immunothérapie avec anti-PD-1, anti-PD-L1, anti-CTLA4 ou tout autre anticorps inhibiteur de point de contrôle immunitaire.
- Épanchement pleural comme seule anomalie radiologique sans épaississement pleural mesurable ni hypertrophie des ganglions médiastinaux.
- Mésothéliome péritonéal, péricardique ou de la tunique vaginale du testicule.
- Diagnostic antérieur d’adénocarcinome de n’importe quel site anatomique au cours des 3 années précédentes, à l’exception d’antécédents d’adénocarcinome de la prostate au cours des 3 années précédentes ; si cancer de la prostate localisé, avec des facteurs de bon pronostic selon la classification de d’Amico (<T2a, score de Gleason ≤6 et PSA ≤10 ng/ml), à condition qu’ils aient été traités selon une modalité curative (chirurgie ou radiothérapie, en l’absence de toute chimiothérapie).Cancer antérieur ou actif au cours des 5 dernières années (à l’exception d’un carcinome in situ du col de l’utérus traité, ou d’un cancer basocellulaire de la peau traité ou non).
- Epanchement pleural non contrôlé nécessitant des ponctions pleurales évacuatrices fréquentes (nécessitant une pleurodèse par thoracoscopie avant inclusion).
- Métastases cérébrales symptomatiques non traitées (sans radiothérapie cérébrale in toto ou radiothérapie cérébrale ablative stéréotaxique préalable ou sans résection chirurgicale). Un délai d’au moins 2 semaines entre la fin de la radiothérapie et le début du traitement par immunochimiothérapie doit être respecté. Les métastases cérébrales asymptomatiques ne nécessitant pas de corticothérapie supérieure à 10 mg de prednisone par jour (ou équivalent) ou de perfusions de mannitol sont autorisées.
- Radiothérapie nécessaire au début du traitement de la tumeur, à l’exception de la radiothérapie palliative osseuse sur une métastase douloureuse ou compressive, ou de la radiothérapie sur les trajets de drain ou ponction thoraciques.
- Antécédents d’immunodéficience primaire, de transplantation d’organe nécessitant un traitement immunosuppresseur, de prise de tout médicament immunosuppresseur dans les 28 jours précédant la date de randomisation, ou antécédents de toxicité sévère (grade 3/4) par mécanisme immunitaire lié à un autre traitement d’immunothérapie pour n’importe quel type de maladie.
- Traitement systémique par corticothérapie à une dose supérieure à 10 mg de prednisone par jour (ou équivalent), dans les 14 jours précédant le début du traitement. Les corticostéroïdes inhalés, nasaux ou thématiques sont autorisés.
- Antécédents de maladie auto-immune active nécessitant un traitement immunosuppresseur systémique, y compris, mais sans s’y limiter, polyarthrite rhumatoïde, myasthénie, hépatite auto-immune, lupus systémique, granulomatose de Wegener, thrombose vasculaire associée au syndrome des anti-phospholipides, syndrome de Sjögren avec maladie pulmonaire interstitielle, syndrome de GuillainBarré récent au cours des 15 dernières années, sclérose en plaques, vascularite ou glomérulonéphrite. Patients atteints de diabète de type I, d’hypothyroïdie, d’une maladie cutanée immunitaire (vitiligo, psoriasis, alopécie) ou d’une polyarthrite rhumatoïde bénigne ne nécessitant pas de traitement systémique immunosuppresseur ou plus de 10 mg de stéroïdes oraux par jour, ou syndrome sec bénin (Sjogren) sans maladie pulmonaire interstitielle, ou antécédents de syndrome de Guillain-Barré depuis plus de 15 ans, totalement réversible et sans séquelles, sans traitement immunosuppresseur systémique au cours des 20 dernières années, peuvent être inclus. Les patients atteints de la maladie de Graves et/ou de psoriasis ne nécessitant pas de traitement systémique au cours des deux dernières années avant la randomisation peuvent être inclus.
- Maladie intestinale inflammatoire active (diverticulose, maladie de Crohn, recto-colite hémorragique, maladie cœliaque) nécessitant un traitement systémique ou toute maladie intestinale chronique grave avec diarrhée incontrôlée.
- Antécédents ou signes actuels d’infection locale ou systémique significative (grade CTCAE ≥2) (par exemple, cellulite, pneumonie, septicémie) nécessitant un traitement antibiotique systémique dans les 2 semaines précédant la première dose du médicament à l’essai.
- Infection active non contrôlée nécessitant un traitement, y compris une tuberculose active. Les antécédents de tuberculose pulmonaire primaire dans la jeunesse ne constituent pas une contre-indication. Les antécédents de tuberculose ne constituent pas une contre-indication à condition que le patient ait été traité pendant au moins 6 mois par un traitement antibiotique antituberculeux.
- Infection non contrôlée par le VIH, le VHB ou le VHC ; ou diagnostic d’immunodéficience liée à une infection chronique ou entraînant une telle infection. Remarques :
- Les patients dont le VIH est connu et dont l’infection est contrôlée (charge virale indétectable et taux de CD4 supérieur à 350, soit spontanément, soit sous un régime antiviral stable) sont autorisés. Pour les patients dont l’infection par le VIH est contrôlée, la surveillance sera effectuée conformément aux pratiques locales.
- Les patients atteints d’une hépatite B connue (HBsAg+) et dont l’infection est contrôlée (PCR de l’ADN du virus de l’hépatite B sérique inférieur à la limite de détection ET traitement antiviral de l’hépatite B) sont autorisés. Les patients dont l’infection est contrôlée doivent faire l’objet d’un contrôle périodique de l’ADN du VHB conformément aux pratiques locales et doivent rester sous traitement antiviral pendant au moins 6 mois après la dernière dose du médicament expérimental.
- Les patients dont on sait qu’ils sont porteurs des anticorps du virus de l’hépatite C (HCV Ab+) et dont l’infection est contrôlée (ARN du HCV indétectable par PCR, soit spontanément, soit en réponse à un traitement anti-HCV antérieur réussi) sont autorisés à participer à l’étude.
- Les patients atteints du VIH ou d’une hépatite doivent être examinés par un spécialiste qualifié (par exemple, un infectiologue ou un hépatologue) qui prend en charge cette maladie avant le début de l’essai et régulièrement pendant toute la durée de leur participation à l’essai.
- Avoir reçu un vaccin vivant dans les 30 jours précédant le début prévu du traitement à l’étude (vaccin vivant ou vivant atténué ayant un potentiel de réplication). Si un patient a l’intention de recevoir un vaccin COVID-19 avant le début du médicament à l’étude, sa participation à l’étude doit être retardée d’au moins une semaine après toute vaccination COVID-19. Pendant la période de traitement, il est recommandé de retarder la vaccination COVID-19 jusqu’à ce que les patients reçoivent et tolèrent une dose régulière du médicament à l’étude. Une dose de vaccin ne doit pas être administrée moins de 48 heures avant ou après l’administration du médicament à l’étude. Les vaccins anti-SARS-CoV2 à ARNm et à vecteur adénovirus sont autorisés.
- Affection générale grave telle qu’une insuffisance cardiaque congestive non contrôlée, une arythmie cardiaque non contrôlée, une cardiopathie ischémique non contrôlée (angine instable ou antécédents d’infarctus du myocarde au cours des 6 mois précédents), des antécédents d’accident vasculaire cérébral au cours des 6 mois précédents, des antécédents de myocardite. Les patients ayant des antécédents cardiaques significatifs, même s’ils sont contrôlés, doivent avoir une FEVG >45 %.
- TnT ou troponine I TnI > 2xULN institutionnel au départ. Les patients dont les taux de TnT ou de TnI se situent entre > 1 et 2xLSN peuvent être inclus si les taux répétés dans les 24 heures sont ≤ 1xULN. Si les taux de TnT ou de TnI sont > 1 à 2xULN dans les 24 heures, le sujet peut subir une évaluation cardiaque et être inclus par l’investigateur sur la base du jugement médical cardiologique dans l’intérêt du patient.
- Hypersensibilité connue aux substances actives ou à l’un des excipients.
- Maladie interstitielle pulmonaire modérée ou sévère préexistante, telle qu’évaluée par le scanner diagnostic et diminution de la TLCO supérieure à 35 % par rapport aux valeurs normales théoriques liées à une telle maladie interstitielle.
- Incapacité à se conformer aux procédures d’étude et/ou de suivi telles qu’estimées par l’investigateur.
- Femmes enceintes ou allaitantes.
- Femmes susceptibles de procréer qui ne veulent pas bénéficier d’une contraception hautement efficace avant le début du traitement, pendant l’étude et pendant au moins 6 mois après la dernière dose. Les mesures contraceptives hautement efficaces comprennent :
- Utilisation régulière d’une contraception hormonale combinée (contenant des œstrogènes et des progestatifs) (orale, intravaginale, transdermique) ou d’une contraception hormonale progestative (orale, injectable, implantable) associée à une inhibition de l’ovulation pendant au moins deux cycles menstruels avant l’inclusion,
- Dispositif intra-utérin ; système intra-utérin de libération d’hormones,
- Occlusion/ligature tubaire bilatérale,
- Partenaire vasectomisé (à condition que le partenaire masculin vasectomisé soit le seul partenaire sexuel de la patiente et que le partenaire vasectomisé ait obtenu une évaluation médicale de la réussite chirurgicale de la procédure), et/ou
- Abstinence sexuelle†. Un test de grossesse et une contraception sont requis. †L’abstinence sexuelle est considérée comme une méthode hautement efficace uniquement si elle est définie comme le fait de s’abstenir de tout rapport hétérosexuel pendant toute la période de risque associée aux médicaments de l’étude. La fiabilité de l’abstinence sexuelle doit être évaluée en fonction de la durée de l’essai clinique et du mode de vie habituel des patientes.
Calendrier prévisionnel
Lancement de l’étude : Mars 2026
Fin estimée des inclusions : Septembre 2027
Nombre de patients à inclure : 126
Information(s) complémentaire(s)
BIO-IFCT-2402
Prélèvements tissulaires
Les biopsies réalisées au moment du diagnostic seront envoyées directement à Mésopath selon les recommandations françaises. Une collection de tissus congelés à partir de biopsies chirurgicales par thoracoscopie (au moment du diagnostic initial du mésothéliome pleural) sera réalisée par le promoteur (IFCT).
Prélèvements sanguins
Des échantillons sanguins seront prélevés à la randomisation, à 24 semaines, 3 mois après la fin de la chimiothérapie (pendant la maintenance par immunothérapie), un an après l’inclusion, à progression. Deux tubes cell-Free DNA de 10ml seront prélevés à chaque point de prélèvement.
Coordonnateur de l'étude
Dr Myriam LOCATELLI-SANCHEZ – Hospices Civils de Lyon
Promoteur de l'étude
Intergroupe Francophone de Cancérologie Thoracique (IFCT)