GORTEC 2024-04 - ILLUMINE
Essai de Phase III, multicentrique, randomisé, en ouvert, évaluant l’ivonescimab seul ou avec le ligufalimab versus pembrolizumab en première ligne de traitement des patients ayant un carcinome épidermoïde de la tête et du cou métastatique ou en récidive
Type d'essai : Académique / Institutionnel
Etat de l'essai : A venir
Situation thérapeutique : Tumeur solide ( Métastatique / Rechute , Localement avancée / Non résécable )
Etablissement(s) participant(s)
Dr Esma SAADA-BOUZID
Dr Benoît CALDERON
Détails de l'essai
Objectif principal
Comparer la survie globale (OS, overall survival) obtenue avec l’ivonescimab associé au ligufalimab à celle obtenue avec le traitement de référence le pembrolizumab et l’OS obtenue avec l’ivonescimab à celle obtenue avec le pembrolizumab chez des patients atteints de CETEC PD-L1-positifs (CPS ≥ 1) R/M 1L.
Objectif(s) secondaire(s)
Comparer l’ORR évalué par le Comité de revue indépendant en aveugle (BIRC, blinded independent review committee) (critère secondaire d’évaluation clé) sur la base des critères RECIST (Response Evaluation Criteria in Solid Tumours) v1.1 obtenu avec l’ivonescimab associé au ligufalimab à celui obtenu avec le pembrolizumab et l’ORR obtenu avec l’ivonescimab à celui obtenu avec le pembrolizumab.
Comparer la PFS évaluée par le BIRC (critère secondaire d’évaluation clé) sur la base des critères RECIST v1.1 obtenue avec l’ivonescimab associé au ligufalimab à celle obtenue avec le pembrolizumab et la PFS obtenue avec l’ivonescimab à celle obtenue avec le pembrolizumab.
Résumé / schéma de l'étude
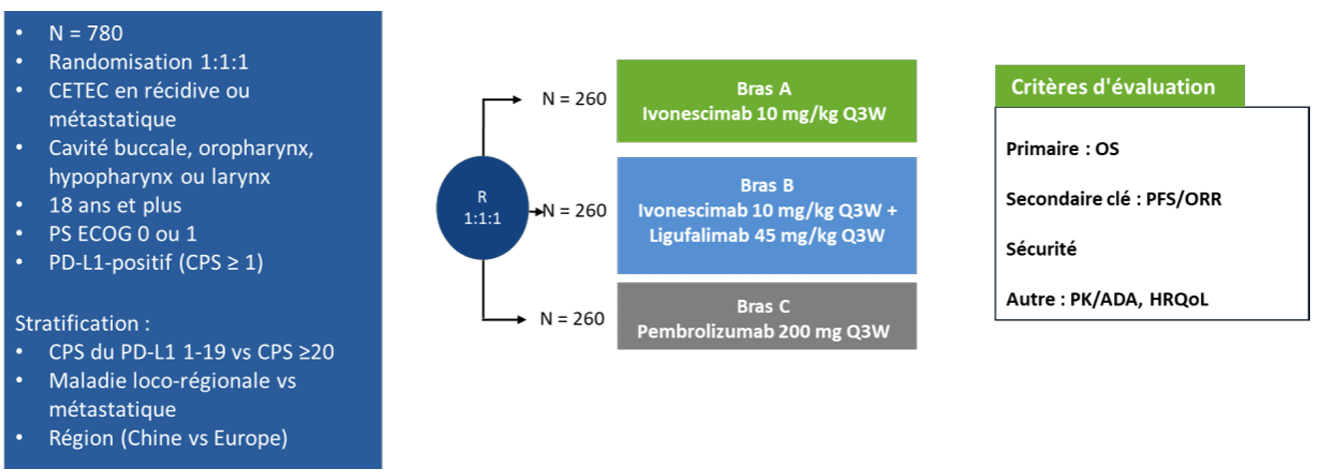
Critère(s) d'inclusion
- Patient capable de donner volontairement son consentement éclairé écrit signé.
- Âge ≥ 18 ans et < 80 ans inclus au moment de l’inclusion.
- Score de statut de performance de l’ECOG (Eastern Cooperative Oncology Group) de 0 ou de 1.
- Survie attendue ≥ 6 mois lors de la randomisation.
- Carcinome épidermoïde des voies aérodigestives supérieures en rechute ou métastatique confirmé histologiquement et/ou cytologiquement, avec une tumeur primitive située initialement ou actuellement dans la cavité buccale, l’oropharynx, l’hypopharynx ou le larynx.
- Les résultats du test concernant le statut de l’HPV, basé sur des échantillons de tissu tumoral, doivent être obtenus avant la randomisation pour les patients atteints d’un cancer oropharyngé.
- Aucun traitement antitumoral systémique antérieur pour le CETEC R/M. Remarque : les patients ayant reçu précédemment une chimiothérapie adjuvante/néoadjuvante à visée curative pour une maladie non métastatique, une radiothérapie ou une radiothérapie définitive associée à une chimiothérapie ou au cetuximab/dirigé contre l’EGFR pour une maladie localement avancée sont éligibles si la progression de la maladie survient plus de 6 mois après la fin du dernier traitement.
- Au moins une lésion mesurable selon les critères RECIST v1.1, ou une lésion mesurable avec progression claire à la radiographie après un traitement local, la lésion devant impérativement permettre des mesures exactes répétées.
- Les tumeurs doivent être PD-L1-positives (CPS ≥ 1) comme confirmé par un test d’immunochimie marqué CE-IVD sur la base d’une évaluation locale avec n’importe quel test validé pour le CETEC dans un laboratoire conforme aux dispositions nationales. La mesure d’expression de la protéine PDL1 peut être effectuée sur la base d’un échantillon tissulaire archivé avant le diagnostic de tumeur R/M ou sur la base d’un échantillon tissulaire obtenu après le diagnostic de tumeur R/M. Les échantillons suivants ne sont pas acceptés : échantillons de cytologie centrifugés provenant du drainage d’un épanchement pleural, de lésions osseuses sans composantes de tissus mous ou d’échantillons de tumeurs osseuses décalcifiées. En outre, un volume suffisant de tissu tumoral doit être archivé pour chaque patient afin de permettre la réalisation ultérieure de 10 lames destinées à un test diagnostique compagnon (CDx, Companion Diagnostic) potentiel.
- Fonction organique adéquate déterminée selon les critères suivants :
- Hématologie (résultats d’analyses de laboratoire satisfaisants obtenus pendant la période de sélection, sans utilisation de composants sanguins dans les 14 jours suivant un traitement de soutien par facteur de croissance cellulaire) :
- Taux absolu de neutrophiles (TAN) ≥ 1,5 x 109/L (1 500/mm3).
- Taux de plaquettes ≥ 100×109/L (100 000/mm3).
- Hémoglobine ≥ 10 g/dL.
- Reins :
- Clairance de la créatinine calculée ≥ 50 mL/min.
- Protéines urinaires ≤ 2+ ou quantification des protéines urinaires sur 24 heures (h) < 1,0 g.
- Foie :
- Bilirubine totale sérique ≤ 1,5 × LSN; pour les patients présentant des métastases hépatiques ou un syndrome de Gilbert confirmé ou suspecté, ≤ 3 × LSN.
- ASAT et ALAT ≤ 2,5 × la LSN ; pour les patients présentant des métastases hépatiques, ASAT et ALAT ≤ 5 × LSN.
- Albuminémie ≥ 28 g/L.
- Fonction de coagulation : Rapport normalisé international (INR, International Normalized Ratio) et/ou temps de céphaline activée (TCA) ≤ 1,5 × LSN. Cela s’applique uniquement aux patients qui ne reçoivent pas de traitement anticoagulant. Les patients sous traitement anticoagulant doivent recevoir une dose stable.
- Hématologie (résultats d’analyses de laboratoire satisfaisants obtenus pendant la période de sélection, sans utilisation de composants sanguins dans les 14 jours suivant un traitement de soutien par facteur de croissance cellulaire) :
- Les patientes en âge de/apte à procréer doivent avoir un résultat négatif au test sérique de grossesse avant la randomisation, ainsi qu’un test urinaire de grossesse négatif dans les 72 heures précédant la première dose. Les participantes ayant des rapports sexuels avec un homme non stérilisé doivent accepter d’utiliser une méthode de contraception hautement efficace à partir du début de la période de sélection et jusqu’à 120 jours après la dernière dose de ligufalimab, d’ivonescimab ou de pembrolizumab. 12.
- Les hommes ayant une partenaire en âge de procréer, enceinte ou allaitante, doivent accepter d’utiliser une contraception barrière (préservatif masculin) pendant toute la durée de la période de traitement et jusqu’à 120 jours après la dernière dose de ligufalimab, d’ivonescimab ou de pembrolizumab. Les hommes ayant une partenaire en âge de procréer doivent s’assurer que celle-ci accepte d’utiliser au moins une forme de contraception hautement efficace pendant toute la durée de la période de traitement et jusqu’à 120 jours après la dernière dose de ligufalimab, d’ivonescimab ou de pembrolizumab.
- Le patient est disposé à et capable de respecter les visites, les protocoles de traitement, les analyses de laboratoire et les autres exigences de l’étude telles que précisées dans le calendrier.
Critère(s) de non-inclusion
- Site tumoral primitif (quelle que soit l’histologie) au niveau du nasopharynx, de la cavité nasale, des sinus, des glandes salivaires, de la thyroïde ou des glandes parathyroïdes, de la peau, ou site primitif inconnu d’origine tissulaire.
- Patient présentant des tumeurs malignes autres qu’un CETEC au cours des 3 ans précédant l’inclusion. Les patients présentant d’autres tumeurs ayant été guéries par un traitement local, telles qu’un carcinome basocellulaire ou épidermoïde cutané, un cancer superficiel de la vessie ou un carcinome in situ du col de l’utérus ou du sein, ne sont pas exclus.
- Participation concomitante à une autre étude clinique, sauf s’il s’agit d’une étude clinique observationnelle non interventionnelle ou de la période de suivi d’une étude interventionnelle ; traitement à l’étude reçu dans les 4 semaines précédant la randomisation.
- Précédent traitement par des agents anti-angiogéniques systémiques.
- Antécédents de ré-irradiation de la tête et du cou pour une maladie en récidive/métastatique.
- Immunothérapie antérieure, y compris par inhibiteurs de point de contrôle immunitaire (par exemple, anticorps anti-PD-1/L1, anti-CTLA-4, anti-TIGIT, anti-LAG3, anti-CD47, anti-SIRPα, etc.), agonistes de point de contrôle immunitaire (par exemple, anticorps ICOS, CD40, CD137, GITR, OX40, etc.), immunothérapie cellulaire, ainsi que tout autre traitement ciblant l’immunité tumorale, notamment les vaccins thérapeutiques antitumoraux et autre traitement adjuvant/néoadjuvant à base d’anti-PD-1.
- Patients présentant des ulcères sur la surface cutanée liés au cancer actuel au cours de la période de sélection, des lésions cutanées superficielles ou saillantes avec une tension superficielle excessive et un risque accru d’ulcération, ou autres patients présentant un risque accru d’ulcération selon l’évaluation de l’investigateur. Patients ayant fait l’objet d’une trachéotomie récente impliquant la tumeur et présentant un risque hémorragique.
- L’imagerie réalisée pendant la période de sélection montre que la tumeur envahit/infiltre les organes importants environnants (tels que la trachée, l’œsophage, et selon l’évaluation par l’investigateur du risque hémorragique) et/ou les gros vaisseaux sanguins du cou (tels que l’artère sous-clavière, l’artère carotide commune, interne et/ou externe, etc.), ou l’investigateur estime que la participation à l’étude pourrait entraîner un risque hémorragique potentiel.
- Présence de métastases du tronc cérébral, de métastases méningées, de métastases ou d’une compression de la moelle épinière, ou d’une maladie leptoméningée.
- Radiothérapie curative de la tête et du cou dans les 6 mois précédant la randomisation. Traitement local palliatif sur des zones autres que la tête et le cou dans les 3 semaines précédant la randomisation ; traitement immunomodulateur non spécifique (tel que l’interleukine, l’interféron, les peptides thymiques, le facteur de nécrose tumorale, etc.) dans les 2 semaines précédant la randomisation, à l’exception de l’IL-11 pour le traitement d’une thrombopénie.
- Présence d’une maladie auto-immune active nécessitant un traitement systémique (par exemple, traitement de fond, corticoïdes, immunosuppresseurs) dans les 2 ans précédant la randomisation. Les thérapies alternatives (par exemple, thyroxine, insuline ou thérapies ciblant les glandes surrénales ou l’hypophyse) ainsi que le traitement substitutif physiologique par corticoïdes en cas d’insuffisance hypophysaire ne sont pas considérés comme des traitements systémiques.
- Maladie inflammatoire chronique de l’intestin avérée active ou antécédents de telle maladie (par exemple, maladie de Crohn, rectocolite hémorragique ou diarrhée chronique).
- Antécédents d’immunodéficience ; précédent test positif aux anticorps anti-VIH ; l’utilisation actuelle à long terme de corticoïdes systémiques ou d’autres immunosuppresseurs est exclue.
- Les patients atteints de tuberculose active connue ou suspectée doivent être exclus après examen clinique ; infection syphilitique active connue.
- Antécédents connus de transplantation d’organe allogénique et de transplantation allogénique de cellules souches hématopoïétiques.
- Pneumopathie inflammatoire/interstitielle non infectieuse antérieure ou actuelle nécessitant un traitement systémique par glucocorticoïdes, ou présence actuelle de maladies pulmonaires incluant, mais sans s’y limiter, la pneumoconiose, la silicose, la pneumonie médicamenteuse, les maladies pulmonaires avec atteinte sévère de la fonction respiratoire, etc.
- Infection sévère dans les 4 semaines précédant la randomisation, incluant notamment les comorbidités nécessitant une hospitalisation, le sepsis ou la pneumonie sévère ; infection non sévère active avec traitement anti-infectieux systémique dans les 2 semaines précédant la randomisation.
- Patients atteints d’hépatite B active (antigène HBs positif et ADN du VHB supérieur à 1 000 copies/ml [200 UI/ml] ou supérieur à la limite inférieure de détection, selon la valeur la plus élevée) ; patients atteints d’hépatite B nécessitant un traitement dirigé contre le virus de l’hépatite B pendant le traitement de l’étude ; patients atteints d’hépatite C active (anticorps anti-VHC positif et ARN du VHC supérieur à la limite inférieure de détection) ; patients infectés à la fois par le VHB et le VHC.
- Chirurgie majeure ou lésion traumatique grave dans les 30 jours précédant la randomisation, ou chirurgie majeure prévue dans les 30 jours suivant la randomisation ; chirurgie locale mineure dans les 3 jours précédant la première dose (la ponction veineuse périphérique, le cathétérisme veineux central et l’implantation d’un accès veineux sont autorisés).
- Présence actuelle de comorbidités médicales non contrôlées, notamment cirrhose décompensée, syndrome néphrotique, troubles métaboliques non contrôlés, maladie ulcéreuse peptique sévèrement active ou gastrite, ou de troubles psychiatriques/conditions sociales qui limiteraient le respect par le patient des procédures de l’étude ou altéreraient sa capacité à fournir un consentement éclairé écrit.
- Angor instable, infarctus du myocarde, insuffisance cardiaque congestive (classification New York Heart Association [NYHA] ≥ grade 2 [voir Annexe 5]) ou maladie vasculaire instable (par exemple, anévrisme aortique à risque de rupture, maladie de Moyamoya) ayant nécessité une hospitalisation dans les 12 mois précédant la randomisation, ou autre insuffisance cardiaque qui pourrait avoir un impact sur l’évaluation de la sécurité du médicament à l’étude (par exemple, arythmies mal contrôlées, ischémie myocardique).
- Antécédents de varices œsophagiennes et gastriques, ulcères sévères, plaies non cicatrisées, fistule abdominale, obstruction gastro-intestinale, abcès intra-abdominal, ou hémorragie gastro-intestinale aiguë dans les 6 mois précédant la randomisation ; exacerbation aiguë d’une bronchopneumopathie chronique obstructive (BPCO) survenue dans le mois précédant la randomisation.
- Tout événement thromboembolique artériel, événement thromboembolique veineux de grade 3 ou plus selon les critères NCI CTCAE version 5.0 (voir Annexe 2), accident ischémique transitoire, accident vasculaire cérébral, poussée hypertensive ou encéphalopathie hypertensive dans les 12 mois précédant la randomisation ; hypertension artérielle actuelle avec pression artérielle systolique ≥ 160 mmHg ou pression artérielle diastolique ≥ 100 mmHg après traitement par des médicaments antihypertenseurs oraux.
- Antécédents de tendance hémorragique ou de troubles de la coagulation sévères ; symptômes ou signes de toute hémorragie tumorale active au cours des 6 mois précédant la randomisation ; présence de symptômes hémorragiques cliniquement significatifs au cours du mois précédant la randomisation, notamment de saignements au niveau de la tête et du cou, de saignements gastro-intestinaux, d’hémoptysie, de saignements nasaux (à l’exclusion de l’épistaxis) ; patients nécessitant un traitement médicamenteux anticoagulant ou antiplaquettaire au long cours.
- Absence de résolution d’une toxicité d’un traitement antinéoplasique antérieur, définie comme une toxicité ne revenant pas à un grade 0 ou 1 selon les critères NCI CTCAE version 5.0 ou au niveau spécifié dans les critères d’inclusion ; une alopécie et une neuropathie périphérique de grade ≤ 2 sont autorisées. Les patients développant une toxicité persistante, pour laquelle il n’est pas attendu d’aggravation après l’administration du médicament à l’étude (par exemple, perte auditive ou toxicité liée à une radiothérapie antérieure telle que xérostomie, dysgueusie, dysphonie, dysphagie, fibrose, œdème cervical, ostéoradionécrose), peuvent être inclus dans l’étude.
- Patient ayant reçu un vaccin vivant dans les 30 jours précédant la randomisation ou chez qui l’administration d’un vaccin vivant est prévue au cours de l’étude.
- Hypersensibilité connue à l’un des composants des médicaments de l’étude ; antécédents connus de réactions sévères d’hypersensibilité à d’autres anticorps monoclonaux.
- Maladie psychiatrique actuelle, abus de substances, d’alcool ou de drogues.
- Femmes enceintes ou qui allaitent.
- Toute maladie antérieure ou actuelle, ou tout traitement qui pourrait fausser les résultats de l’étude, avoir un impact sur la participation du patient ou ne pas être dans le meilleur intérêt du patient.
- Présence d’une maladie locale ou systémique non causée par la tumeur maligne, ou d’une maladie ou de symptômes secondaires à la tumeur, qui pourraient entraîner un risque médical accru ou une incertitude dans l’évaluation du risque (par exemple, réaction leucémoïde due à la tumeur [numération des globules blancs > 20×10⁹/l], cachexie [perte de poids mensuelle de plus de 10 % au cours des 3 mois précédant la sélection]).
- Les personnes privées de liberté, faisant l’objet d’une mesure de protection juridique et/ou non affiliées à un système de sécurité sociale, doivent être exclues.
- Une masse tumorale importante et/ou une progression rapide de la maladie qui nécessiteraient le recours à la chimiothérapie dans le cadre du traitement de première ligne.
Calendrier prévisionnel
Lancement de l’étude : Avril 2026
Fin estimée des inclusions : Avril 2030
Nombre de patients à inclure : 390
Coordonnateur de l'étude
Dr Aurore VOZY – AP-HP
Promoteur de l'étude
Groupe d’Oncologie Radiothérapie Tête Et Cou (GORTEC)