Prodige 89 - ALICE
Etude randomisée intégrée de Phase II/III comparant durvalumab et tremelimumab +/- GEMOX en chimiothérapie intra-artérielle hépatique dans les carcinomes hépatocellulaires à charge tumorale élevée
Phase : II / III
Type d'essai : Académique / Institutionnel
Etat de l'essai : Ouvert
Situation thérapeutique : Tumeur solide ( Métastatique / Rechute , Localement avancée / Non résécable )
Type d'essai : Académique / Institutionnel
Etat de l'essai : Ouvert
Situation thérapeutique : Tumeur solide ( Métastatique / Rechute , Localement avancée / Non résécable )
Etablissement(s) participant(s)
Dr Xavier ADHOUTE
Dr Laurent MINEUR
Détails de l'essai
Objectif principal
Phase II : Comparer l’efficacité entre les deux bras en termes de Taux de Réponses objectives (ORR) selon RECIST v1.1.
Phase III : Comparer l’efficacité entre les deux bras en termes de Survie Globale (OS).
Objectif(s) secondaire(s)
Evaluer la sécurité de la combinaison immunothérapie / CIAH.
Comparer l’efficacité entre les deux bras en termes de :
o Survie sans progression (PFS) selon RECIST 1.1 et mRECIST
o L’ORR selon RECIST 1.1 (pour la phase III)
o La survie globale (OS) (pour la phase II)
Comparer la qualité de vie (HR-QoL) pendant l’étude.
Résumé / schéma de l'étude
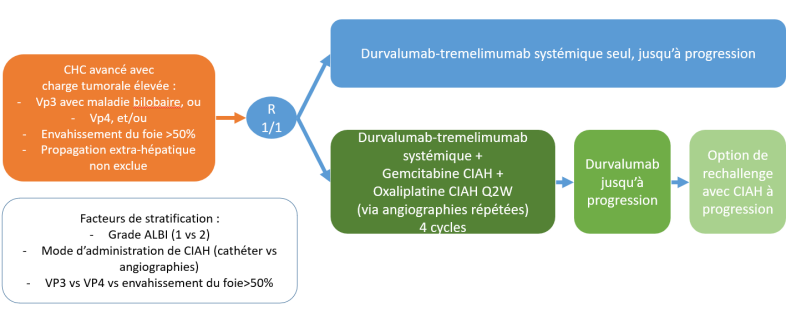
Critère(s) d'inclusion
- Âge ≥ 18 ans.
- Patient présentant un carcinome hépatocellulaire (CHC), de stade avancé ou non résécable diagnostiqué soit à l’histologie soit par les critères radiologiques EASL, si aucune biopsie n’a pu être réalisée en toute sécurité.
- Charge tumorale élevée, définie par l’un de ces trois critères : (i) PVTT Vp4, (ii) PVTT Vp3 avec envahissement tumoral bilobaire et/ou (iii) envahissement du foie >50% (selon l’évaluation de l’investigateur). L’extension extra-hépatique est autorisée.
- Classification Child-Pugh A, de stade B ou C selon la classification Barcelona Clinic Liver Cancer classification (BCLC) de l’hépatocarcinome.
- Score de performance ECOG 0 ou 1.
- Espérance de vie d’au moins 12 semaines.
- Poids corporel > 30 kg.
- Au moins une lésion cible mesurable à l’inclusion, non préalablement irradiée. L’évaluation tumorale par scanner ou IRM doit être réalisée dans les 28 jours avant randomisation.
- Fonction adéquate des organes et de la moelle, traduite par les valeurs de laboratoire suivantes :
- Hémoglobine ≥ 9 g/dL
- Plaquettes ≥ 100 × 109/L,/L
- Neutrophiles ≥ 1.0 × 109/L
- Clairance de la créatinine > 40 mL/min (méthodes Cockcroft ou MDRD)
- ASAT /ALAT ≤5x limite normale supérieure (LNS)
- Bilirubine sérique ≤1.5 x LNS.
- International normalised ratio (INR) < 2.3
- Les femmes en âge de procréer doivent utiliser un moyen de contraception hautement efficace pendant toute la durée de l’étude, et jusqu’à 90 jours après la dernière dose de durvalumab en monothérapie ou de durvalumab et de trémélimumab en association, pendant au moins 15 mois après la fin du traitement par oxaliplatine, pendant au moins 6 mois après la fin du traitement par gemcitabine, et présenter un test de grossesse négatif urinaire ou sérique dans les 7 jours avant la première administration de traitement. En cas de test urinaire, il doit posséder une sensibilité très élevée.
- Les hommes non stériles doivent accepter d’utiliser une méthode contraceptive hautement efficace pendant toute la durée de l’étude et jusqu’à ≥90 jours après la dernière dose de durvalumab en monothérapie ou de durvalumab et de tremelimumab en association, pendant au moins 12 mois après la fin du traitement à l’oxaliplatine, pendant au moins 3 mois après la fin du traitement à la gemcitabine. Un homme est défini comme stérile lorsqu’une azoospermie a été constatée lors d’un examen de sperme et considérée comme preuve définitive d’infertilité. Les hommes ayant un faible nombre de spermatozoïdes (connue sous les termes « sous fertilité » ou fertilité relative) ne doivent pas être considérés comme stériles dans le cadre de cette étude.
- Les hommes et femmes participant à cette étude doivent accepter de ne pas faire de don de sperme ou d’ovocyte pendant la durée du traitement et jusqu’à 180 jours après l’arrêt du traitement.
- Le patient doit avoir signé et daté un formulaire de consentement éclairé écrit avant toute procédure, analyse ou prélèvement spécifique à l’essai. Lorsque le patient est physiquement incapable de donner son consentement écrit, une personne de confiance de son choix, indépendante de l’investigateur ou du promoteur, peut confirmer par écrit le consentement du patient.
- Le patient est désireux et capable de se conformer au protocole pendant toute la durée de l’essai, y compris en ce qui concerne le traitement et les visites prévues, ainsi que les examens, y compris le suivi.
- Les patients doivent être affiliés à un système de sécurité sociale.
Critère(s) de non-inclusion
- Traitement systémique antérieur (qu’il s’agisse d’immunothérapie, d’antiangiogéniques, de chimiothérapie ou d’une combinaison de ces traitements).
- Traitement antérieur par Chimiothérapie Intra-Artérielle Hépatique.
- CHC fibrolamellaire, sarcomatoïde ou mélange de CHC et de cholangiocarcinome.
- Antécédent d’encéphalopathie hépatique au cours des 6 derniers mois ou nécessitant un traitement curatif ou préventif (par ex, utilisation de lactulose ou de rifaximin pour l’encéphalopathie hépatique).
- Saignement gastrointestinal actif ou lors des 6 derniers mois (par ex, varices œsophagiennes ou saignement d’ulcère). Note : pour les participants ayant un antécédent de saignement de plus de 6 mois, ou jugés à haut risque de varices œsophagiennes par l’investigateur (thrombose du tronc porte principal inclus), il sera nécessaire de réaliser une endoscopie dans les 3 mois précédant l’inclusion et une thérapie endoscopique adéquate selon les standards institutionnels.
- Toute toxicité résiduelle de grade ≥2 selon le NCI CTCAE d’un traitement anticancéreux antérieur, à l’exception de l’alopécie, du vitiligo et des valeurs de laboratoires dont les seuils sont définis dans les critères d’inclusion.
- Les patients avec une neuropathie de Grade ≥2 seront évalués au cas par cas après consultation avec l’investigateur.
- Les patients avec une toxicité irréversible dont on ne peut raisonnablement envisager qu’elle soit augmentée par le durvalumab ou le tremelimumab pourront être inclus après validation d’un investigateur.
- Tout autre traitement anti-cancéreux, y compris les chimiothérapies, les thérapies biologiques ou hormonales. Un traitement hormonal non lié au cancer (par ex, thérapie hormonale de substitution) est acceptable.
- Antécédents de maladie auto-immune active (y compris les maladies inflammatoires de l’intestin [par ex, colites ou maladie de Crohn], diverticulites [à l’exception de la diverticulose], lupus érythémateux systémique, syndrome de Sarcoïdose, ou syndrome de Wegener [granulomatose avec polyangéite, maladie de Graves, arthrite rhumatoïde, hypophysite, uvéite, etc.]). Les cas ci-dessous sont des exceptions :
- Patients avec vitiligo ou alopécie
- Patients avec hypothyroïdisme (par ex, suivant un syndrome d’Hashimoto) stables sous hormone de substitution
- Toute maladie cutanée chronique ne nécessitant pas de traitement systémique
- Les patients dont la maladie n’a pas été active au cours des 5 dernières années peuvent être inclus, après validation d’un investigateur formé à l’étude
- Patients avec maladie cœliaque contrôlée par un régime alimentaire seul
- Antécédents de maladie pulmonaire interstitielle, de pneumonie non infectieuse ou de maladies non contrôlées, y compris la fibrose pulmonaire, les maladies pulmonaires aiguës.
- Antécédent de carcinomatose leptoméningée.
- Hépatite infectieuse active, positivité aux anticorps de l’hépatite C (HCV), de l’antigène de surface de l’hépatique B (HBsAg) ou de l’anticorps HBV (anti-HBc), à l’inclusion. Les participants avec une infection HBV ancienne ou guérie (définie par la présence d’anti-HBc et l’absence de HBsAg) sont éligibles. Les participants positifs aux anticorps HCV sont éligibles seulement si leur PCR pour l’ARN HCV est négative.
- Résultat positif connu au test du Virus de l’Immunodéficience Humaine (VIH) (Anticorps VIH 1/2 positifs) ou tuberculose active (évaluation clinique incluant les antécédents, l’examen physique et les résultats radiologiques, ou dépistage de la tuberculose selon les pratiques locales).
- Maladie intercurrente non contrôlée, incluant les infections en cours ou actives, l’insuffisance cardiaque congestive symptomatique, l’hypertension non contrôlée, l’angine de poitrine non stable, l’arythmie cardiaque, la maladie pulmonaire interstitielle, les conséquences gastrointestinales chroniques graves de la diarrhée.
- Intervalle de QT corrigé selon la formule de Fridericia (QTcF) ≥470 ms, calculé à partir de 3 ECGs (réalisés à 5 minutes d’écart chacun).
- Prise de traitement immunosuppresseur en cours ou dans les 14 jours avant la première administration de durvalumab et de tremelimumab. Exceptions à cette contre-indication : stéroïdes topiques inhalés par voie nasale, ou injection locale de stéroïdes (par ex, injection intra-articulaire); corticostéroïdes systémiques à dose physiologique, n’excédant pas 10 mg/jour de prednisone ou équivalent; stéroïdes en prémédication pour les réactions d’hypersensibilité (par ex, prémédications avant un CT scan).
- Vaccin vivant reçu dans les 30 jours avant la première dose du traitement à l’étude.
- Antécédent de transplantation allogénique, ou patient éligible à la transplantation, le patient ne doit pas être éligible à une greffe de foie.
- Procédure chirurgicale majeure (selon le jugement de l’investigateur) dans les 28 jours avant la première administration de traitement à l’étude. Note : les chirurgies locales de lésions isolées, à visée palliative, sont acceptables.
- Présence d’un cancer antérieur actif au cours des 5 années précédant l’inclusion à l’exception des cancers curables localement considérés comme guéris ou réséqués avec succès, tels que les cancers basocellulaires ou épidermoïdes de la peau, le cancer superficiel de la vessie, les cancers gastriques, le carcinome in situ de la prostate, du col de l’utérus, ou les carcinomes mammaires. Tout traitement oncologique concomitant est interdit pendant la phase de traitement.
NB : radiothérapie dans les 28 jours avant la première dose du traitement à l’étude. - Allergie ou hypersensibilité connue à l’un des traitements à l’étude, ou à l’un des excipients de ces traitements.
- Les femmes enceintes ou allaitantes, ainsi que les hommes ou femmes en âge de procréer et refusant d’employer un contraceptif efficace du screening jusqu’à 90 jours après la dernière dose de durvalumab en monothérapie, ou jusqu’à 180 jours après la dernière administration de la combinaison durvalumab et tremelimumab.
- Participation à une autre étude clinique thérapeutique dans les 30 jours avant l’inclusion.
- Traitement antérieur ou randomisation antérieure dans une étude clinique incluant le durvalumab et/ou le tremelimumab, quel qu’ait été le bras de traitement assigné.
- Participation concomitante à une autre étude clinique, sauf les études observationnelles (non-interventionnelles) et sauf pendant la période de suivi des études interventionnelles.
- Patients privés de libertés ou sous tutelle ou curatelle.
- Patients incapables de respecter le protocole pour des raisons géographiques, sociales ou psychologiques, ou maladie psychiatrique/condition sociale qui limiterait la compliance aux exigences de l’étude, augmenterait le risque d’occurrence des AE ou compromettrait la capacité du patient à émettre un consentement éclairé par écrit.
- Troubles ioniques tels que :
- Potassium < 3,5 mEq/L (< 3,5 mmol/L)
- Magnesium < 1,8 mg/dL (< 0,70 mmol/L)
- Concentration de calcémie totale < 8.8 mg/dL (< 2.20 mmol/L).
Calendrier prévisionnel
Lancement de l’étude : Novembre 2025
Fin estimée des inclusions : Septembre 2029
Nombre de patients à inclure : 196
Coordonnateur de l'étude
Pr Julien EDELINE – Centre Eugène Marquis – CLCC Rennes
Promoteur de l'étude
UNICANCER
Dernière mise à jour le 17 février 2026