GRACIE
Etude de phase II évaluant l’ivonescimab en association avec la chimiothérapieen première et deuxième ligne de traitement chez des patients atteints d’adénocarcinome gastrique et gastro-œsophagien avancé ou métastatique
Type d'essai : Académique / Institutionnel
Etat de l'essai : Ouvert
Situation thérapeutique : Tumeur solide ( Métastatique / Rechute , Localement avancée / Non résécable )
Etablissement(s) participant(s)
Dr Christelle De La FOUCHARDIERE
Détails de l'essai
Objectif principal
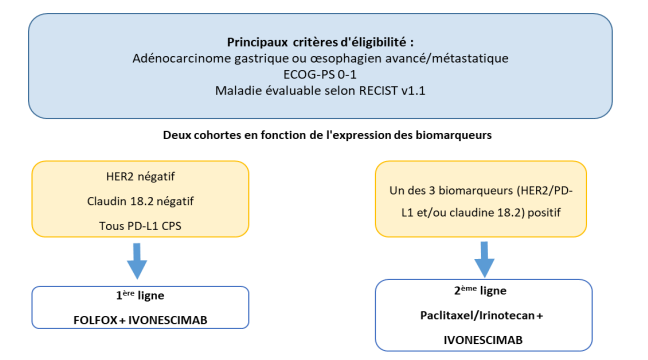
Evaluer l’efficacité de l’ivonescimab associé à la chimiothérapie chez les patients atteints de GEAC avancé ou métastatique, avec ou sans biomarqueur actionnable (HER2/PD-L1/claudin18.2) en termes de taux de réponse objective (TRO) tel qu’évalué par la relecture centralisée selon RECIST v1.1.
Objectif(s) secondaire(s)
Evaluer l’efficacité de l’ivonescimab en termes de :
– TRO tel qu’évalué par l’investigateur selon RECIST v1.1
– Durée de la réponse (DoR) selon RECIST V1.1
– Survie Sans Progression (SSP)
– Survie Globale (SG)
– Délai de détérioration de l’état de performance ECOG
Evaluer la toxicité et le profil de sécurité de l’ivonescimab associé à la chimiothérapie.
Evaluer la qualité de vie liée à la santé (QdV) à l’aide du QLQ-C30 et du QLQ-OG25 chez les patients traités par l’ivonescimab en association avec la chimiothérapie.
Résumé / schéma de l'étude
Critère(s) d'inclusion
- Avoir signé un formulaire de consentement éclairé avant toute procédure spécifique à l’essai.
Note: Si le patient est physiquement incapable de donner son consentement écrit, une personne de confiance de son choix, indépendante de l’investigateur ou du promoteur, peut confirmer le consentement du patient par écrit. - Cancer gastrique (CG) ou adénocarcinome cancéreux de la jonction œsogastrique (CJOG) histologiquement ou cytologiquement prouvé.
- Maladie métastatique ou localement avancée non résécable (stade IV).
- Présence d’au moins une lésion mesurable évaluée par l’investigateur selon RECIST v1.1.
- Statut de performance de l’Eastern Cooperative Oncology Group (ECOG) de 0 ou 1.
- Âge ≥18 ans.
- Pour les patients sans biomarqueur actionnable (HER2 et Claudin 18.2-négatifs) à l’exception de PD-L1, pas de traitement antérieur pour une maladie avancée (cohorte 1). Pour les patients présentant au moins un des biomarqueurs actionnables suivants (PD-L1 CPS≥1, et/ou HER2-positif, et/ou Claudin 18.2-positif), une seule ligne de traitement préalable pour une maladie avancée (cohorte 2).
- Fonction hématologique adéquate : numération absolue des neutrophiles ≥1,5 × 10⁹/L, numération plaquettaire ≥100 × 10⁹/L et hémoglobine ≥9 g/dL.
Note: Aucune transfusion sanguine ou thérapie par facteur de croissance ne doit être effectuée dans les 7 jours précédant l’analyse hématologique. - Fonction rénale adéquate : clairance de la créatinine estimée ≥50 ml/min selon la formule Cockcroft-Gault, ou valeur du débit de filtration glomérulaire estimé (DFG) ≥50 ml/min selon l’équation de la Chronic Kidney Disease Epidemiology Collaboration (CKD-EPI), et protéines urinaires < 2+ ou quantification des protéines urinaires sur 24 heures < 1,0 g.
- Fonction hépatique adéquate : taux de bilirubine totale ≤1,5 × LSN (≤3 x LSN pour les patients présentant des métastases hépatiques ou un syndrome de Gilbert confirmé/suspecté), et taux d’aspartate aminotransférase (AST) et d’alanine aminotransférase (ALT) ≤2,5 × LSN (AST et ALT ≤5 x LSN en cas de métastases hépatiques documentées).
- Coagulation : temps de prothrombine (TP) ou rapport international normalisé (INR) ≤ 1,5 x LSN, et temps de céphaline (PTT) ou temps de céphaline activée (aPTT) ≤ 1,5 × LSN (sauf si les anomalies ne sont pas liées à une coagulopathie). Cela s’applique uniquement aux patients qui ne sont pas sous anticoagulation thérapeutique. Les patients recevant une anticoagulation thérapeutique doivent avoir une dose stable.
- Les femmes en âge de procréer ayant des rapports sexuels avec un partenaire masculin non stérilisé et les hommes non stérilisés ayant des rapports sexuels avec une partenaire féminine en âge de procréer doivent accepter d’utiliser une méthode de contraception efficace pendant toute la durée de leur participation à l’essai et, le cas échéant, après la fin du traitement de l’étude (120 jours après la dernière dose d’ivonescimab). Les hommes doivent également accepter de ne pas faire de don de sperme et les femmes doivent accepter de ne pas faire de don d’ovocytes pendant la période spécifiée.
- Les femmes en âge de procréer doivent avoir un test de grossesse sérique négatif dans les 3 jours précédant l’inclusion et un test de grossesse urinaire négatif le jour de la première dose, avant l’administration du traitement.
- Volonté et capacité de respecter le protocole pendant toute la durée de l’étude, y compris les visites programmées, le plan de traitement, les tests de laboratoire et les autres procédures de l’étude.
- Affiliation au système de sécurité sociale (ou équivalent).
Critère(s) de non-inclusion
- Cancer antérieur ou concomitant dont le site primaire ou l’histologie est distincte de celle du cancer gastro-œsophagien dans les 2 ans précédant l’inclusion dans l’étude, à l’exception du cancer in situ du col de l’utérus traité de manière curative, du cancer de la peau sans mélanome et des tumeurs superficielles de la vessie [Ta (tumeur non invasive), Tis (carcinome in situ) et T1 (invasion de la lamina propria)].
- Patients présentant une instabilité microsatellitaire élevée (MSI-H) ou un mismatched repair disease (dMMR).
- Apport entéral < 1500 kcal/j et/ou perte de poids > 15% du poids corporel total au cours des 6 derniers mois.
- Toxicités du traitement précédent non résolues à un grade ≤ 1 (selon la version 5.0 du National Cancer Institute – Common terminology criteria for adverse events [NCI-CTCAE v5.0]) avant le début du traitement, à l’exception de l’alopécie.
- Interventions chirurgicales majeures ou traumatismes graves dans les 4 semaines précédant le début du traitement, ou projets d’interventions chirurgicales majeures dans les 4 semaines suivant la première dose (selon la décision de l’investigateur). Interventions locales mineures (à l’exception du cathétérisme veineux central et de l’implantation d’un port) dans les 3 jours précédant le début du traitement.
- Antécédents de tendances hémorragiques ou de coagulopathie et/ou symptômes ou risques hémorragiques cliniquement significatifs dans les 4 semaines précédant l’inclusion, y compris mais non limité à :
- Hémoptysie (définie comme l’expectoration de ≥ 0,5 cuillère à café de sang frais ou de petits caillots de sang)
Note: une hémoptysie passagère associée à une bronchoscopie diagnostique est autorisée. - Saignement nasal /épistaxis (un écoulement nasal sanglant est autorisé),
- L’utilisation actuelle d’anticoagulants prophylactiques ou à pleine dose ou d’agents antiplaquettaires à des fins thérapeutiques qui n’est pas stable avant le début du traitement n’est pas autorisée. L’utilisation d’anticoagulants à pleine dose est autorisée tant que le rapport international normalisé (INR) ou le temps de thromboplastine partielle activée se situe dans les limites thérapeutiques selon les normes médicales de l’établissement d’inscription.
- Hémoptysie (définie comme l’expectoration de ≥ 0,5 cuillère à café de sang frais ou de petits caillots de sang)
- Hypertension mal contrôlée avec une pression artérielle systolique répétée ≥ 150 mmHg ou une pression artérielle diastolique ≥ 100 mmHg après un traitement antihypertenseur oral.
- Antécédents de maladies graves avant l’inclusion, en particulier :
- Angor instable, infarctus du myocarde, insuffisance cardiaque congestive (classification de la New York Heart As-sociation ≥ grade 2) ou maladie vasculaire instable (par exemple, anévrisme aortique présentant un risque de rupture, maladie de Moyamoya) ayant nécessité une hospitalisation dans les 12 mois précédant l’inclusion, ou autre déficience cardiaque susceptible d’affecter l’évaluation de la sécurité du médicament à l’étude (par exemple, arythmies mal contrôlées, ischémie myocardique).
- Antécédents de varices œsophagiennes et gastriques, d’ulcères graves, de plaies qui ne cicatrisent pas, de fistules abdominales, d’abcès intra-abdominaux ou d’hémorragies gastro-intestinales aiguës dans les 6 mois précédant l’inclusion.
- Antécédents d’événement thromboembolique artériel, d’événement thromboembolique veineux de grade 3 ou plus selon la norme NCI-CTCAE v5.0, d’accident ischémique transitoire, d’accident vasculaire cérébral, de crise hypertensive ou d’encéphalopathie hypertensive dans les 12 mois précédant l’inclusion.
- Exacerbation aiguë d’une bronchopneumopathie chronique obstructive dans les 4 semaines précédant l’inclusion.
- Antécédents de perforation du tractus gastro-intestinal et/ou de fistule, antécédents d’obstruction gastro-intestinale (y compris obstruction intestinale incomplète nécessitant une alimentation parentérale), résection intestinale étendue (colectomie partielle ou résection étendue de l’intestin grêle) dans les 6 mois précédant l’inclusion.
- L’imagerie réalisée au cours de la période de dépistage montre que le patient a :
- Preuve radiologiquement documentée de l’invasion ou de l’encapsulation de vaisseaux sanguins majeurs par le cancer.
- Preuve radiographique de cavitation intra-tumorale.
- Preuve d’un risque hémorragique plus élevé sur les prothèses.
- Toute thérapie immunosuppressive pendant plus de 7 jours (c’est-à-dire corticostéroïdes >10mg ou dose équivalente) dans les 14 jours précédant le début prévu de la thérapie de l’étude. La thérapie physiologique de remplacement des corticostéroïdes en cas d’insuffisance surrénale ou hypophysaire n’est pas considérée comme une forme de traitement systémique.
- Maladie auto-immune active ayant nécessité un traitement systémique au cours des deux dernières années (c’est-à-dire corticoïdes ou médicaments immunosuppresseurs). Le traitement de substitution (par ex. thyroxine, insuline) est autorisé Maladie auto-immune active ou antécédents de maladie auto-immune ou de déficit immunitaire, y compris, mais sans s’y limiter, myasthénie grave, myosite, hépatite auto-immune, lupus érythémateux disséminé, polyarthrite rhumatoïde, maladie inflammatoire de l’intestin, syndrome des anticorps antiphospholipides, granulomatose de Wegener, syndrome de Sjögren, syndrome de GuillainBarré ou sclérose en plaques, à l’exception des cas suivants :
- Les patients ayant des antécédents d’hypothyroïdie d’origine auto-immune et qui prennent une hormone de remplacement de la thyroïde sont éligibles à l’étude.
- Les patients atteints de diabète sucré de type 1 contrôlé et soumis à un régime d’insuline sont éligibles à l’étude.
- Les patients atteints d’eczéma, de psoriasis, de lichen simplex chronique ou de vitiligo avec des manifestations dermatologiques uniquement (par exemple, les patients atteints d’arthrite psoriasique sont exclus) sont éligibles pour l’étude si toutes les conditions suivantes sont remplies :
- L’éruption doit couvrir moins de 10 % de la surface corporelle.
- La maladie est bien contrôlée au départ et ne nécessite que des corticoïdes topiques peu puissants.
- Absence d’exacerbations aiguës de l’affection sous-jacente nécessitant l’administration de psoralen et de rayons ultraviolets A, de méthotrexate, de rétinoïdes, d’agents biologiques, d’inhibiteurs de la calcineurine par voie orale ou de corticostéroïdes puissants ou par voie orale au cours des 12 derniers mois.
- Antécédents connus d’hypersensibilité à l’ivonescimab et aux autres IMP prévus (fluorouracile, oxaliplatine, acide folinique, irinotécan et paclitaxel) ou à l’un de leurs excipients.
- En fonction du traitement prévu :
- Régime FOLFOX :
- Traitement récent ou concomitant par la brivudine
- Infection potentiellement grave
- Patients en mauvais état nutritionnel
- Vaccins vivants atténués
- Neuropathie sensorielle périphérique avec déficience fonctionnelle avant le premier traitement
- Hypokaliémie, hypomagnésémie, hypocalcémie
- Intervalle QT/QTc supérieur à 450 msec pour les hommes et à 470 msec pour les femmes sur l’ECG d’inclusion.
- Irinotecan :
- Utilisation concomitante avec le millepertuis
- Maladie inflammatoire chronique de l’intestin
- Vaccins vivants atténués
- Régime FOLFOX :
- Greffe de moelle osseuse allogénique antérieure ou greffe d’organe solide antérieure.
- Antécédents connus ou signes de maladie pulmonaire interstitielle.
- Patient présentant une infection non contrôlée par le virus de l’immunodéficience humaine (VIH) (anticorps VIH 1/2 positifs) (patient présentant une charge virale indétectable (PCR ARN VIH) et un taux de CD4 supérieur à 350, soit spontanément, soit sous traitement antiviral stable).
- Infection chronique par le virus de l’hépatite B ou C (si le statut de l’hépatite ne peut être obtenu à partir des dossiers médicaux, un nouveau test est nécessaire)
- En cas de traitement prévu par le fluorouracile, un déficit complet prouvé en dihydropyrimidine déshydrogénase (DPD).
- Toute condition qui, de l’avis de l’investigateur, rendrait indésirable la participation du sujet à l’essai ou compromettrait le respect du protocole.
- Femmes enceintes ou allaitantes.
- Participation à un autre essai thérapeutique dans les 30 jours précédant l’entrée dans l’étude. La participation à un essai d’observation est acceptable.
- Patients ne voulant pas ou ne pouvant pas se conformer au suivi médical requis par l’essai pour des raisons géographiques, familiales, sociales ou psychologiques.
- Personnes privées de liberté ou placées sous protection ou tutelle.
Calendrier prévisionnel
Lancement de l’étude : Janvier 2026
Fin estimée des inclusions : Janvier 2027
Nombre de patients à inclure : 88
Coordonnateur de l'étude
Dr Christelle DE LA FOUCHARDIERE – Institut Paoli Calmettes – CLCC Marseille
Promoteur de l'étude
UNICANCER