IFCT 2404 - COMET
Etude randomisée COntrôlée comparant le Tepotinib au traitement standard chez des patients avec un Cancer Bronchique Non à Petites Cellules de stade avancé ayant une mutation de l’exon 14 de MET
Type d'essai : Académique / Institutionnel
Etat de l'essai : Ouvert
Situation thérapeutique : Tumeur solide ( Localement avancée / Non résécable , Métastatique / Rechute )
Cibles / marqueurs : METex14
Etablissement(s) participant(s)
Détails de l'essai
Objectif principal
Comparer l’efficacité du tepotinib au traitement choisi par l’investigateur chez des patients ayant un CBNPC METex14 après une ligne d’immunothérapie et/ou de chimiothérapie à base de platine mesurée par la survie sans progression (PFS).
Objectif(s) secondaire(s)
Evaluer la qualité de vie sous tepotinib ou traitement choisi par l’investigateur mesurée par l’évolution entre l’inclusion et la semaine 6 en utilisant le questionnaire QLQ-C30-
LC29 ->1ère analyse hiérarchique → effectuée uniquement si l’analyse de la PFS est statistiquement significative.
Evaluer l’efficacité du tepotinib ou du traitement choisi par l’investigateur mesuré par la survie globale (OS).
-> 2ème analyse hiérarchique → effectuée uniquement si l’analyse de la PFS et de la qualité de vie sont statistiquement significatives.
Evaluer l’efficacité du tepotinib ou du traitement choisi par l’investigateur mesurée par :
o Le taux de réponse objective selon RECIST 1.1.
o La durée de réponse.
Evaluer l’efficacité du premier traitement ultérieur mesurée par :
o La PFS2.
o Le temps jusqu’au prochain traitement ou décès (TNT-D).
Evaluer la sécurité sous tepotinib ou sous le traitement choisi par l’investigateur : incidence, nature et gravité des évènements indésirables (CTCAE v5.0).
Evaluer l’observance du traitement mesurée par :
o Le nombre de cycles dans chaque bras.
o Le nombre de modification de dose et d’interruption du traitement.
Résumé / schéma de l'étude
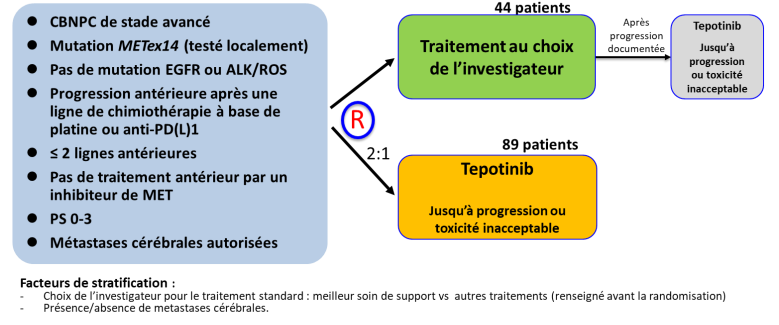
Critère(s) d'inclusion
- Consentement éclairé, écrit et signé :
– Les patients doivent avoir signé et daté le formulaire de consentement éclairé écrit approuvé par le comité d’éthique en accord avec le cadre juridique et institutionnel.
Il doit avoir été signé avant que des procédures liées au protocole ne faisant pas partie de la prise en charge standard des patients soient réalisées.
– Les patients doivent être disposés et capables de respecter le calendrier des visites, du traitement et des analyses de laboratoire. - CBNPC confirmé histologiquement.
- Présence d’une mutation de l’exon 14 de MET (testé localement). La détection de la mutation METex14 doit être effectuée sur un échantillon tissulaire si celui-ci est disponible. S’il n’y a pas d’échantillon tissulaire disponible, la détection de la mutation METex14 sur une biopsie liquide est autorisée. En cas de doute sur la nature de la mutation, un avis du promoteur avant inclusion est recommandé.
- Progression de la maladie après au moins une ligne de traitement incluant soit une chimiothérapie à base de platine, soit un agent anti-PD(L)1, soit les deux.
- Ne pas avoir reçu plus de deux lignes de traitement antérieures.
- ECOG Performance Status PS 0-3.
- Les métastases cérébrales sont autorisées. Si un traitement local immédiat est nécessaire, l’inclusion est possible dès que celui-ci sera terminé.
- Stade IIIB ou IIIC non irradiable ou stade IV (8ème classification TNM, UICC 2015)
- Age ≥ 18 ans.
- Fonction biologique adéquate :
- Clairance de la créatinine ≥ 30 ml/min
- Neutrophiles ≥ 1500 / mm3
- Plaquettes ≥ 100 000 / mm3
- Hémoglobine ≥ 8 g/dL
- Enzymes hépatiques < 3 x LNS (< 5 x LNS en cas de métastases hépatiques)
- Bilirubine totale ≤1,5 x LNS (< 5 x LNS en cas de syndrome de Gilbert ou < 3 x LNS en cas de métastases hépatiques).
- Les majeurs protégés peuvent être inclus s’ils sont capables de prendre des décisions concernant leur traitement médical conformément au jugement de tutelle.
- Pour les femmes en âge de procréer (y compris les femmes ayant subi une ligature des trompes), un test de grossesse sérique est à effectuer dans les 14 jours avant le C1J1 et doit
être négatif. - Les femmes en âge de procréer doivent rester abstinentes (s’abstenir de rapports hétérosexuels) ou utiliser des méthodes contraceptives dont le taux d’échec est inférieur à 1 % par an pendant la période de traitement et pendant au moins 6 mois après la dernière dose du traitement de l’étude. Les femmes doivent s’abstenir de faire un don d’ovocyte durant
cette même période. Une femme est considérée comme étant en âge de procréer si elle est post-ménarche, n’a pas atteint un état post-ménopausique (≥ 12 mois continus d’aménorrhée sans cause identifiée autre que la ménopause) et n’a pas subi de stérilisation chirurgicale (ablation des ovaires ou de l’utérus). Les méthodes contraceptives dont le taux d’échec est inférieur à 1 % par an incluent la ligature bilatérale des trompes, la stérilisation masculine, l’utilisation correcte de contraceptifs hormonaux inhibant l’ovulation, les dispositifs intrautérins à libération d’hormones et les dispositifs intra-utérins en cuivre. Les méthodes contraceptives hormonales doivent être complétées avec une méthode de barrière avec spermicide. La fiabilité de l’abstinence sexuelle doit être évaluée en fonction de la durée de l’étude clinique et du mode de vie préféré et habituel du patient. L’abstinence périodique (par exemple, les méthodes calendaires, d’ovulation, symptothermiques ou de postovulation) et le retrait ne sont pas des méthodes de contraception acceptables. - Les hommes ayant une partenaire féminine en âge de procréer ou une partenaire féminine enceinte doivent rester abstinents ou utiliser un préservatif pendant la période de traitement et pendant au moins 6 mois après la dernière dose du traitement à l’étude afin d’éviter d’exposer l’embryon. Les hommes doivent s’abstenir de donner leur sperme pendant cette même période. La fiabilité de l’abstinence sexuelle doit être évaluée en fonction de la durée de l’étude clinique et du mode de vie préféré et habituel du patient. L’abstinence périodique (par exemple, les méthodes calendaires, d’ovulation, symptothermiques ou de postovulation) et le retrait ne sont pas des méthodes de contraception acceptables.
- Patient bénéficiant d’une couverture par une assurance maladie nationale.
Critère(s) de non-inclusion
- Traitement antérieur avec un inhibiteur de MET (y compris le crizotinib).
- Présence d’une autre altération oncogénique connue (y compris les mutations EGFR, HER2, KRAS, BRAF ou les fusions ALK, ROS1, RET). En cas de détection d’une autre altération
oncogénique, l’inclusion doit être discutée avec le promoteur. - ECOG Performance Status PS 4.
- Hypersensibilité connue au tepotinib ou à ses excipients.
- Antécédents de cancer datant de moins de 3 ans ou cancer actif, à l’exception de ceux présentant un risque négligeable de métastases ou de décès, ou de ceux traités de manière
curative. Si un patient ne remplit pas ce critère mais que l’investigateur considère que la balance bénéfices/risques est en faveur de son inclusion dans l’étude, veuillez contacter
l’IFCT. - Incapacité à respecter les procédures de l’étude ou de suivi.
- Femmes enceintes, allaitantes ou en cours d’allaitement.
- Maladie, dysfonctionnement métabolique, résultat d’examen physique ou de laboratoire clinique permettant de suspecter raisonnablement une maladie ou un état qui contre-indique l’utilisation d’un médicament expérimental, qui peut affecter l’interprétation des résultats ou qui peut exposer le patient à un risque élevé de complications liées au traitement.
- Antécédents de fibrose pulmonaire idiopathique ou de pneumopathie active au scanner thoracique lors du screening. Les antécédents de pneumopathie radique dans le champ
d’irradiation (fibrose) sont autorisés.
Calendrier prévisionnel
Lancement de l’étude : Juillet 2025
Fin estimée des inclusions : Juillet 2027
Nombre de patients à inclure : 133
Coordonnateur de l'étude
Pr Alexis CORTOT – CHU Lille
Promoteur de l'étude
Intergroupe Francophone de Cancérologie Thoracique (IFCT)