Prodige 78 - SAFIR-ABC10 Precision Medicine
Thérapie d’entretien ciblée versus traitement standard dans le cancer biliaire avancé : un essai de phase III international, randomisé, contrôlé, en ouvert
Type d'essai : Académique / Institutionnel
Etat de l'essai : Ouvert
Situation thérapeutique : Tumeur solide ( Métastatique / Rechute , Localement avancée / Non résécable )
Etablissement(s) participant(s)
Dr Ludovic EVESQUE
Dr Laetitia DAHAN
Dr Hervé PERRIER
Dr Nicolas BARRIERE
Dr Simon LAUNAY
Dr May MABRO
Détails de l'essai
Objectif principal
Evaluer si le traitement d’entretien avec des MTT pour des cibles « exploitables » après 4 cycles de 1L-SoC est supérieur à la poursuite de 1L-SoC en termes de survie sans progression (PFS – progression-free survival).
Objectif(s) secondaire(s)
Evaluer l’efficacité de :
– Oncologie de précision vs 1L-SoC chez les patients ABC présentant des altérations moléculaires tumorales « établies ».
– Chaque MTT (ou combinaison de MTT).
Evaluer la faisabilité du profilage moléculaire dans un essai académique multinational.
Comparer la qualité de vie des patients traités avec des MTT vs le 1L-SoC.
Evaluer la sécurité des thérapies ciblées.
Etudier la valeur pronostique des altérations moléculaires, quelle que soit la randomisation.
Résumé / schéma de l'étude
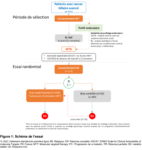
Bras expérimental : Thérapie moléculaire ciblée assignée par le MTB, à administrer selon la posologie indiquée dans le tableau ci-dessus.
Bras contrôle : Poursuite du 1L-SoC – CISGEM ± durvalumab* 4 cycles de cisplatine 25 mg/m2 IV suivis de gemcitabine 1000 mg/m2 IV aux jours 1 et 8 Q3W (CISGEM) ± durvalumab 1500 mg IV à jour 1 Q3W, puis durvalumab seul 1500 mg IV Q4W après l’arrêt du CISGEM.
*Durvalumab peut être prescrit à la discrétion des Investigateurs dans les territoires où il est disponible.
Critère(s) d'inclusion
Phase de sélection
- Signé un formulaire de consentement éclairé écrit avant toute procédure spécifique à l’essai (Consentement #1).
A noter : Si le patient est physiquement incapable de fournir son consentement écrit, une personne de confiance de son choix, indépendante de l’investigateur ou du promoteur, peut confirmer le consentement du patient par écrit. - Cholangiocarcinome intrahépatique ou périhilaire/distal ou carcinome de la vésicule biliaire histologiquement prouvé (carcinome ampullaire exclu).
- Maladie de novo ou récurrente, localement avancée (non-résécable) ou métastatique.
- Disponibilité d’un échantillon archivé de tissu tumoral primaire ou métastatique (congelé ou FFPE) ou capable de subir une biopsie pour obtenir un échantillon de tissu malin.
- Âgé ≥ 18 ans.
- Statut de performance ECOG de 0 ou 1.
- Espérance de vie estimée > 3 mois.
- Candidat à la thérapie 1L-SoC ou dont le 1ier cycle de thérapie 1L-SoC a été initié.
- Affilié à un système de sécurité sociale ou en possession d’une assurance maladie privée équivalente (conformément aux conditions du pays).
Essai randomisé
- Signé un formulaire de consentement éclairé écrit avant toute procédure spécifique à l’essai (Consentement #2).
A noter : Si le patient est physiquement incapable de fournir son consentement écrit, une personne de confiance de son choix, indépendante de l’investigateur ou du promoteur, peut confirmer le consentement du patient par écrit. - Profil moléculaire montrant que la tumeur abrite au moins une altération moléculaire ciblée par un des MTT dans le portefeuille de l’étude (tel que déterminé par le MTB)
- Maladie contrôlée (stable ou répondeur) après 4 cycles de 1L-SoC, par rapport à une évaluation de la maladie faite avant le traitement, telle qu’évaluée par l’investigateur
- Statut de performance ECOG de 0 ou 1
- Présence d’au moins une lésion évaluable selon RECIST v1.1, ou réponse complète (CR) à 4 Cycles de 1L-SoC.
- Fonction adéquate de la moelle osseuse : numération absolue des neutrophiles (ANC) ≥ 1,5 × 10⁹/L, numération plaquettaire ≥ 100 × 10⁹/L et hémoglobine ≥ 9 g/dL.
- Fonction hépatique adéquate : taux de bilirubine totale ≤ 1,5 × la limite supérieure de la normale (LSN) (taux de bilirubine totale ≤ 3.0 LSN si le patient présente un syndrome de Gilbert documenté), et taux d’aspartate aminotransférase (ASAT) et d’alanine aminotransférase (ALAT) ≤ 2,5 × LSN (ASAT et ALAT ≤ 5 x LSN en cas d’atteinte hépatique tumorale documentée).
- Fonction rénale adéquate : clairance estimée de la créatinine ≥ 60 mL/min selon la formule de Cockcroft-Gault Fonction cardiaque adéquate : fraction d’éjection ventriculaire gauche ≥ 50 % à la baseline, déterminée soit par échocardiogramme, soit par acquisition multiple (MUGA).
- Drainage biliaire adéquat, sans signe d’infection en cours.
- Les hommes, et les femmes en âge de procréer, doivent accepter d’utiliser une contraception adéquate pendant la durée de leur participation à l’essai et selon les recommandations après la fin du traitement de l’étude. Les hommes doivent également accepter de ne pas donner de sperme et les femmes doivent accepter de ne pas donner d’ovocytes pendant la période spécifiée.
- Les femmes en âge de procréer doivent avoir un test de grossesse sérique négatif effectué dans les 3 jours précédant la date de randomisation.
- Volonté et capacité de se conformer au protocole pendant toute la durée de l’étude, y compris aux visites programmées, au plan de traitement, aux tests de laboratoire et à d’autres procédures de l’étude.
- Affilié à un système de sécurité sociale ou en possession d’une assurance maladie privée équivalente (conformément aux conditions du pays).
Passage du bras contrôle à la thérapie MTT
- Maladie évolutive documentée pendant ou après l’arrêt du 1L-SoC.
- ECOG PS de 0 ou 1.
- Fonction adéquate de la moelle osseuse : Neutrophiles ≥ 1,5 × 10⁹/L, numération plaquettaire ≥ 100 × 10⁹/L et hémoglobine ≥ 9 g/dL.
- Fonction hépatique adéquate : taux de bilirubine totale ≤ 1,5 × LSN sauf si le patient présente un syndrome de Gilbert documenté, et taux d’AST et d’ALT ≤ 2,5 × LSN (ASAT et ALAT ≤ 5 LSN en cas de métastases hépatiques documentées).
- Fonction rénale adéquate : clairance estimée de la créatinine ≥ 45 mL/min selon la formule de Cockcroft-Gault.
- L’obstruction des voies biliaires a été soulagée.
- Les hommes, et les femmes en âge de procréer, doivent accepter d’utiliser une contraception adéquate pendant la durée de leur participation à l’essai et selon les recommandations après la fin du traitement de l’étude. Les hommes doivent égalementaccepter de ne pas donner de sperme et les femmes doivent accepter de ne pas donner d’ovocytes pendant la période spécifiée.
- Les femmes en âge de procréer doit avoir un test de grossesse sérique négatif effectué dans les 3 jours avant le début du traitement ciblé.
Critère(s) de non-inclusion
Phase de sélection
- Contre-indication au 1L-SoC.
- Patients candidats à une thérapie locorégionale.
- Contre-indication à la biopsie tumorale en l’absence d’échantillon de tissu tumoral archivé.
- Chimiothérapie palliative antérieure. Capécitabine adjuvante autorisée si terminée ≥ 183 jours avant l’entrée dans l’étude.
- Avoir déjà reçu plus d’1 cycle de thérapie 1L-SoC.
- Traitement antérieur avec l’un des MTT étudiés dans SAFIR-ABC10.
- Tumeur maligne concomitante (autres qu’ABC), à l’exception des carcinomes in situ du col de l’utérus et des carcinomes basocellulaires ou épidermoïdes de la peau, correctement traités. Les survivants du cancer, qui ont suivi un traitement potentiellement curatif pour une tumeur maligne antérieure, et qui n’ont aucun signe de cette maladie depuis 5 ans ou plus, et qui sont considérés comme présentant un risque négligeable de récidive, sont éligibles pour l’essai.
- Toute condition qui, de l’avis de l’investigateur, rend indésirable la participation du sujet à l’essai ou qui compromettrait le respect du protocole.
- Femmes enceintes ou qui allaitent.
- Patients refusant ou incapables de se conformer au suivi médical requis par l’essai pour des raisons géographiques, familiales, sociales ou psychologiques.
- Personnes privées de liberté ou placées sous tutelle/curatelle ou en garde à vue.
Essai randomisé
- Progression de la maladie survenant à tout moment avant la randomisation, ou toxicité ayant conduit à l’arrêt de la chimiothérapie 1L-SoC avant que 4 cycles complets aient été délivrés.
- Toxicités du 1L-SoC non résolues au grade ≤ 1 (selon la version 5.0 du National Cancer Institute – Common terminology criteria for adverse events [NCI-CTCAE v5.0]) avant la randomisation, à l’exception de l’alopécie.
- Contre-indication ou hypersensibilité connue au MTT pour l’altération moléculaire constatée chez le patient, ou tout composant de sa formulation.
A noter : Pour les patients présentant plusieurs altérations cibles, la contre-indication à un MTT ne justifiera pas l’exclusion du patient s’il peut recevoir le MTT pour une autre cible. - Cancers avec instabilité microsatellite élevée (MSI-H) ou déficience de réparation des mésappariements (dMMR).
- Chirurgie majeure dans les 4 semaines suivant la randomisation.
- Radiothérapie dans les 7 jours suivant la randomisation.
- Métastases du système nerveux central (SNC) non traitées, métastases symptomatiques du SNC ou radiothérapie pour les métastases du SNC dans les 4 semaines suivant le début du traitement à l’étude. Les métastases cérébrales stables et traitées sont autorisées (définies comme des sujets qui ne prennent plus de stéroïdes et antiépileptiques et qui sont neurologiquement stables sans signe de progression radiologique pendant au moins 4 semaines au moment du dépistage).
- Maladie cardiovasculaire cliniquement significative (infarctus aigu du myocarde récent, insuffisance cardiaque congestive traitée [2 ou plus sur l’échelle de classification fonctionnelle de la New York Heart Association], événements thromboemboliques ou cérébrovasculaires récents [dans les 12 semaines, sauf s’ils sont liés à un cathéter à demeure], syndrome du QT prolongé connu).
- Pathologies cardiorespiratoires où l’hyperhydratation est contre-indiquée.
- Manifestation d’acouphènes et/ou de perte auditive depuis le début du traitement par cisplatine.
- Maladie leptoméningée connue. Si une maladie leptoméningée a été rapportée par imagerie sur l’IRM de baseline, mais n’est pas suspectée cliniquement par l’investigateur, le sujet doit être exempt de symptôme neurologique.
- Tumeur maligne concomitante (autre qu’ABC), à l’exception d’un carcinome in situ du col de l’utérus et d’un carcinome basocellulaire ou épidermoïde de la peau, correctement traités. Les survivants du cancer, qui ont suivi un traitement potentiellement curatif pour une tumeur maligne antérieure, et qui n’ont aucun signe de cette maladie depuis 5 ans ou plus et qui sont considérés comme présentant un risque négligeable de récidive, sont éligibles pour l’essai.
- Traitement concomitant par la phénytoïne à usage prophylactique lorsque celui-ci ne peut pas être remplacé par un autre traitement.
- Infection active connue au virus de l’hépatite B ou au virus de l’hépatite C ou au virus de l’immunodéficience humaine (VIH) ou syndrome d’immunodéficience acquise connu.
- Toute condition qui, de l’avis de l’investigateur, rend indésirable la participation du sujet à l’essai ou qui compromettrait le respect du protocole.
- Femmes enceintes ou allaitantes.
- Participation à un autre essai thérapeutique dans les 30 jours précédant l’entrée dans l’étude. La participation à un essai observationnel est acceptable.
- Patients refusant ou incapables de se conformer au suivi médical requis par l’essai pour des raisons géographiques, familiales, sociales ou psychologiques.
- Personnes privées de liberté ou placées sous tutelle/curatelle ou en garde à vue.
Critères de non-inclusion supplémentaires applicables uniquement aux patients éligibles à des MTT spécifiques
Patients destinés à recevoir des thérapies orales :
- Incapacité ou refus d’avaler des pilules.
- Antécédents de syndrome de malabsorption ou autre affection susceptible d’interférer avec l’absorption entérale. Par exemple, inflammation intestinale active (maladie de Crohn ou colite ulcéreuse, etc…) nécessitant un traitement immunosuppresseur.
Futibatinib :
- Antécédents et/ou signes actuelles de l’un des troubles suivants :
a. Altération de l’homéostasie calcium-phosphore non liée à la tumeur et considérée comme cliniquement significative par l’investigateur. La concentration de phosphate sanguin doit être ≤ LSN lors de l’examen initial.
b. Minéralisation/calcification ectopique, y compris, mais sans s’y limiter, aux tissus mous, aux reins, à l’intestin ou au myocarde et aux poumons, considérée comme cliniquement significative par l’investigateur
c. Trouble rétinien ou cornéen confirmé par un examen rétinien/cornéen et considéré comme cliniquement significatif par l’investigateur. - Traitement concomitant avec des inhibiteurs forts du CYP3A/P-gp ou des inducteurs forts ou modérés du CYP3A/P-gp lorsque ceux-ci ne peuvent pas être remplacés par un autre traitement.
Ivosidenib :
- Patients ayant des antécédents de torsade de pointes.
- Traitement concomitant avec la digoxine lorsque celle-ci ne peut être substituée à un autre traitement.
- Patients avec un intervalle QT corrigé de la fréquence cardiaque (selon la formule de Fridericia) (QTcF) ≥ 450 msec ou d’autres facteurs qui augmentent le risque d’allongement de l’intervalle QT ou d’événements arythmiques (par exemple, insuffisance cardiaque, hypokaliémie, antécédents familiaux de syndrome d’intervalle QT long).
- Traitement concomitant par des inducteurs forts du CYP3A4 ou du dabigatran lorsque ceux-ci ne peuvent pas être remplacés par un autre traitement.
- Traitement concomitant avec des médicaments connus pour prolonger l’intervalle QTc ou des inhibiteurs modérés ou forts du CYP3A4 lorsque ceux-ci ne peuvent pas être remplacés par un autre traitement.
- Antécédents familiaux de mort subite ou d’arythmie ventriculaire polymorphe.
- Hypokaliémie, hypomagnésémie ou hypocalcémie lorsqu’elles ne peuvent être corrigées par une supplémentation.
Zanidatamab:
- Traitement par anthracyclines dans les 90 jours précédant la première dose de zanidatamab et/ou charge totale à vie supérieure à 360 mg/m2 d’Adriamycine® ou équivalent.
- Utilisation de corticostéroïdes administrés à des doses équivalentes à > 15 mg par jour de prednisone dans les 2 semaines suivant la première dose de zanidatamab, sauf autorisation contraire de l’investigateur coordinateur. Les corticostéroïdes topiques, oculaires, intra-articulaires, intranasaux et/ou inhalés sont autorisés.
- QTcF > 470 msec.
- Antécédents d’infarctus du myocarde ou d’angor instable dans les 6 mois précédant l’inclusion, taux de troponine compatibles avec un infarctus du myocarde ou maladie cardiaque cliniquement significative, telle qu’une arythmie ventriculaire nécessitant un traitement, une hypertension non contrôlée ou tout antécédent d’insuffisance cardiaque congestive symptomatique.
- Pancréatite aiguë ou chronique non contrôlée ou maladie hépatique de Child-Pugh C.
- Maladie pulmonaire infiltrante cliniquement significative non liée à des métastases pulmonaires.
- Antécédents d’hypersensibilité potentiellement mortelle aux anticorps monoclonaux ou aux protéines recombinantes.
Neratinib & trastuzumab
- Patients atteints d’insuffisance hépatique sévère (Child-Pugh C).
- Co-administration avec les produits médicamenteux, qui sont de puissants inducteurs de l’isoforme CYP3A4/P-gp du cytochrome P450, tels que la carbamazépine, la phénytoïne (antiépileptiques), le millepertuis (Hypericum perforatum) ou la rifampicine (antimycobactérien).
- Patients souffrant de dyspnée au repos en raison de complications d’une tumeur maligne avancée ou de comorbidités.
- Hypersensibilité aux protéines murines.
- Pneumopathie active dans les 90 jours suivant l’initiation du trastuzumab ou antécédents connus de maladie pulmonaire interstitielle.
Encorafenib & binimetinib
- Patients ayant des antécédents ou des signes actuels d’occlusion veineuse rétinienne ou de facteurs de risque d’occlusion veineuse rétinienne (par exemple, glaucome non contrôlé ou antécédents d’hyperviscosité ou de syndrome d’hypercoagulabilité)
- Patients atteints de troubles neuromusculaires concomitants associés à un taux élevé de creatine phosphokinase (>LSN)
- Patients présentant une hypokaliémie, une hypomagnésémie ou une hypocalcémie (c’està-dire potassium sérique, magnésium ou calcium < limite normale inférieure)
- Patients avec un QTcF ≥ 450 msec pour les hommes ou ≥ 470 msec pour les femmes.
- Utilisation actuelle ou prévue d’un inhibiteur puissant du CYP3A4
Passage du bras contrôle à la thérapie MTT
- Contre-indication au MTT pour l’altération moléculaire constatée chez le patient (y compris les critères d’exclusion supplémentaires spécifiques au MTT énumérés ci-dessus).
- Hypersensibilité antérieure connue au MTT pour l’altération moléculaire trouvée chez le patient, ou à tout composant de leur formulation.
- Traitement avec tout agent antinéoplasique dans le cadre de l’entretien ou de la deuxième intention, après l’arrêt du 1L-SoC.
- Toxicités du 1L-SoC non résolues au grade ≤ 2 (NCI-CTCAE v5.0) avant le début du MTT à l’exception de l’alopécie.
- 1L-SoC interrompu > 6 semaines avant le début du traitement croisé MTT.
- Toute condition qui, de l’avis de l’investigateur, rend indésirable la participation du sujet à l’essai ou qui compromettrait le respect du protocole.
- Femme enceinte ou allaitante.
Calendrier prévisionnel
Lancement de l’étude : Mai 2024
Fin estimée des inclusions : Juin 2027
Nombre de patients à inclure : 800 patients sélectionnés / 159 patients inclus
Coordonnateur de l'étude
Dr David MALKA
Institut Mutualiste Montsouris
Promoteur de l'étude
UNICANCER