RIBOLARIS / SOLTI-1911
Ribociclib et endocrinothérapie en traitement néoadjuvant et adjuvant dans le cancer du sein avec statut des récepteurs des oestrogènes positif (RE+) et HER2 négatif (HER2-) à risque clinique élevé
Type d'essai : Académique / Institutionnel
Etat de l'essai : Ouvert
Situation thérapeutique : Tumeur solide ( Adjuvant , Néoadjuvant )
Etablissement(s) participant(s)
TMSCB / chimiothérapie / immunothérapie / thérapies ciblées / A1 / RTA / RTB / curiethérapie / colo / côlon / intestin / anus / canal anal / voie biliaire / B1 / rectum / foie / pancréas / œsophage / estomac / A4 / prostate / rein / vessie / uretère / urètre / testicule / pénis / B4 / A5 / endomètre / col utérin / utérus / vulve / vagin / B5 / ovaire / A6 / sein / séno / A7 / mélanome / cutané / peau / dermatologie
Dr Alexandre TASSIN De NONNEVILLE
TMSCA / chimiothérapie / immunothérapie / thérapies ciblées / RTA / RTB / curiethérapie
Dr Julien GRENIER
Détails de l'essai
Objectif principal
Evaluer l’innocuité et le long terme efficacité d’un traitement non chimio chez les patients biologiquement répondeurs au traitement néoadjuvant ribociclib et létrozole.
Résumé / schéma de l'étude
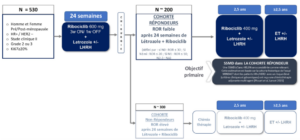
Critère(s) d'inclusion
- Formulaire de consentement éclairé signé avant toute procédure spécifique à l’étude. Les patients doivent être désireux et capable de se conformer au protocole pendant toute la durée de l’étude, y compris visites programmées, plan de traitement, tests de laboratoire et autres procédures d’étude. Remarque : les patients candidats en France doivent être affiliés à un système de sécurité sociale (ou équivalent).
- Hommes (≥ 18 ans) ou femmes pré-ménopausées (≥ 40 ans) ou femmes post-ménopausées. Les patients préménopausés/hommes recevront des agonistes de la LHRH 2 semaines avant C1D1 et pendant traitement. Le statut post-ménopausique est défini comme : Âge ≥ 60 ans ou âge < 60 ans et 12 mois d’aménorrhée plus hormone folliculo-stimulante (FSH) et les taux plasmatiques d’œstradiol (E2) dans la plage post-ménopausique par le laboratoire local évaluation ou ovariectomie bilatérale préalable (≥ 7 jours avant le jour 1 du traitement).
- Carcinome mammaire invasif histologiquement confirmé, confirmé par le pathologiste, avec toutes les caractéristiques suivantes :
- Stade clinique II (septième édition de l’AJCC) qui comprend cT1cN1cM0, cT2cN0cM0, cT2cN1cM0 et cT3cN0cM0.
- ER-positif/HER2-négatif selon les dernières directives ASCO/CAP évalué localement, cellules tumorales > 10 % de coloration ER, cancer du sein de grade 2 ou 3.
- Indice Ki-67 par analyse locale de ≥ 20 % sur du tissu mammaire non traité. Noter: Les tumeurs multifocales et multicentriques sont autorisées si elles sont considérées comme cliniques stade II selon la septième édition de l’AJCC. La biopsie de toutes les lésions n’est pas nécessaire.
- Cancer du sein éligible à une chirurgie primaire.
- Biopsie de noyau FFPE de pré-traitement disponible (tru-cut) évaluable pour PAM50 ou possibilité pour en obtenir un. Les exigences minimales en matière d’échantillon sont d’avoir au moins 1 cylindre tumoral avec une surface tissulaire minimale de 10 mm2 de tissu, contenant au moins 10 % de cellules tumorales et avoir suffisamment de tissu pour faire au moins 2 coupes de 10 μm chacune (la qualité de l’échantillon doit être approuvé au niveau central avant l’inclusion).
- Avoir un statut de performance Eastern Cooperative Oncology Group (ECOG) de 0 à 1. L’évaluation de l’ECOG doit être effectuée dans les 14 jours précédant la date d’inscription.
- Fonction hématologique, rénale et hépatique adéquate, comme suit :
- Nombre absolu de neutrophiles (ANC) ≥ 1,5 x 109 /L.
- Numération plaquettaire ≥100 x 109 /L.
- Hémoglobine ≥ 10 g/dL.
- Phosphatase alcaline (AP) ≤ 2,5 x LSN.
- Bilirubine totale < LSN. Les patients atteints du syndrome de Gilbert connu peuvent être inscrits avec bilirubine totale ≤ 3 x LSN ou bilirubine directe ≤ 1,5 x LSN.
- Alanine aminotransférase (ALT) et aspartate aminotransférase (AST) < 2,5 x LSN.
- Créatinine sérique ≤ 1,5 mg/dL ou clairance de la créatinine calculée ≥ 60 mL/min (Équation de Cockcroft-Gault).
- Potassium, calcium total (corrigé pour l’albumine sérique), magnésium et sodium dans les limites normales de l’établissement ou corrigées dans les limites normales avec suppléments avant la première dose du médicament à l’étude.
- Participants masculins : Un participant masculin doit accepter d’utiliser un moyen de contraception tel que détaillé à l’annexe 1 de ce protocole pendant la période de chimiothérapie adjuvante (seulement cohorte non-répondeur) et pour au moins 21 jours, correspondant au temps nécessaire pour éliminer tout traitement à l’étude plus 120 jours supplémentaires (un cycle de spermatogenèse) après la dernière dose de chimiothérapie et s’abstenir de donner du sperme pendant cette période. Après la fin du traitement d’essai, les patientes doivent utiliser une contraception efficace conformément aux directives locales.
- Participantes féminines : Une participante est éligible à participer si elle n’est pas enceinte, l’allaitement, et au moins une des conditions suivantes s’applique (voir annexe 1) :
- Pas une femme en âge de procréer (WOCBP) telle que définie à l’annexe 1.
- OU Un WOCBP qui accepte de suivre les conseils contraceptifs de l’annexe 1 pendant la période de traitement et pendant au moins 21 jours (correspondant au temps nécessaire pour éliminer tout traitement à l’étude) plus 30 jours (un cycle menstruel) pour l’étude traitements à risque de génotoxicité après la dernière dose du traitement à l’étude. Après la fin du traitement d’essai, les patients doivent utiliser une contraception efficace selon les directives locales.
Critère(s) de non-inclusion
- Tout traitement antérieur pour le cancer du sein invasif primaire. Létrozole ou autres médicaments utilisés pendant la préservation de la fonction ovarienne sont autorisés s’ils sont administrés après biopsie de base.
- Cancer du sein inopérable.
- Les patientes atteintes d’un cancer du sein de stade I, III ou IV ne sont pas éligibles. Mise en scène de base à documenter l’absence de maladie métastatique n’est pas nécessaire, mais est recommandé comme déterminé par la pratique institutionnelle (chez les patients où il peut y avoir un suspicion de maladie avancée, par exemple grosses tumeurs, lymphe axillaire cliniquement positive ganglions, signes et symptômes). S’ils sont effectués, les rapports de ces examens doivent être disponible. Type d’examen pour la stadification, c’est-à-dire radiographie, échographie, scintigraphie osseuse, tomodensitométrie, IRM, et/ou PET-CT, est à la discrétion de l’investigateur.
- Cancer du sein invasif bilatéral.
- Patients ayant subi une biopsie du ganglion sentinelle avant le traitement à l’étude.
- Incapacité ou refus d’avaler des pilules.
- Syndrome de malabsorption ou autre affection susceptible d’interférer avec l’absorption entérique de médicaments à l’étude.
- Participation à une étude expérimentale antérieure dans les 30 jours précédant l’inscription ou dans les 5 demi-vies du produit expérimental, selon la plus longue.
- Patient avec un score de Child-Pugh B ou C.
- Le patient a une maladie cardiaque active ou des antécédents de dysfonctionnement cardiaque, y compris du suivant :
- Antécédents de syndromes coronariens aigus (incluant infarctus du myocarde, angine de poitrine, pontage aortocoronarien, angioplastie coronarienne ou pose de stent) ou péricardite symptomatique dans les 12 mois précédant le dépistage.
- Antécédents d’insuffisance cardiaque congestive documentée (New York Heart Association classification fonctionnelle III-IV).
- Cardiomyopathie documentée.
- Le patient a une fraction d’éjection ventriculaire gauche (FEVG) < 50 %, déterminée par Scan ou échocardiogramme (ECHO) d’acquisition multiple Gated (MUGA).
- Arythmies cardiaques cliniquement significatives (par exemple, tachycardie ventriculaire), bloc de branche gauche complet, bloc AV de haut grade (par exemple bloc bifasciculaire, Mobitz type II et bloc AV du troisième degré).
- Syndrome du QT long ou antécédents familiaux de mort subite idiopathique ou Syndrome QT ou l’un des éléments suivants : facteurs de risque de torsades de pointe (TdP), y compris hypokaliémie non corrigée ou hypomagnésémie, antécédents d’insuffisance cardiaque ou antécédents de bradycardie significative/symptomatique.
- QTc > 500 msec ou anomalie de conduction au cours des 12 derniers mois.
- Lors du dépistage ECG 12 dérivations, l’un des paramètres cardiaques suivants : bradycardie (fréquence cardiaque au repos < 50), tachycardie (fréquence cardiaque au repos > 90), intervalle PR > 220 ms, intervalle QRS > 109 ms ou intervalle QTcF ≥ 450 ms (en utilisant la méthode de Fridericia correction).
- Hypertension non contrôlée (pression artérielle systolique > 160 mmHg ou <90 mmHg et/ou diastolique >100 mmHg).
- Infection active nécessitant des antibiotiques intraveineux (IV).
- Antécédents de pneumonite de toute cause.
- Événements thromboemboliques antérieurs non attribuables à une cause déclenchante claire.
- Infection connue par le virus de l’immunodéficience humaine (VIH).
- Toute autre maladie, dysfonctionnement pulmonaire actif ou non contrôlé, métabolisme dysfonctionnement, résultat d’examen physique ou résultat de laboratoire clinique donnant suspicion raisonnable d’une maladie ou d’un état qui contre-indique l’utilisation d’un médicament expérimental, qui peut compromettre le respect du protocole, qui peut affecter l’interprétation des résultats ou exposer les patients à un risque élevé de complication du traitement.
- Lésion traumatique importante dans les 3 semaines précédant le début du traitement de l’étude.
- Intervention chirurgicale majeure (à l’exclusion des interventions mineures telles que la biopsie des ganglions lymphatiques, biopsie au trocart tumoral, aspiration à l’aiguille fine ou ovariectomie bilatérale) dans les 3 semaines avant le début du traitement à l’étude ou pas complètement récupéré de tout effet secondaire de procédures précédentes.
- Toute condition psychologique, familiale, sociologique ou géographique potentiellement entravant le respect du protocole d’étude et du calendrier de suivi.
- Les patients ayant des antécédents de malignité ne sont pas éligibles, sauf pour les cas suivants :
- Les patientes ayant des antécédents de malignité autres que le cancer du sein invasif sont éligibles s’ils sont indemnes depuis au moins 5 ans et sont jugés par le que l’investigateur présente un faible risque de récidive de cette tumeur maligne.
- Les patients atteints des cancers suivants sont éligibles, même s’ils sont diagnostiqués et traités au cours des 5 dernières années : carcinome canalaire in situ du sein, cancer du col de l’utérus cancers de la peau in situ et non métastatiques non mélanomateux.
- La thérapie de remplacement des œstrogènes s’est arrêtée moins de 2 semaines avant le début du traitement.
- Hypersensibilité connue à l’un des excipients du ribociclib, du létrozole, de la goséréline ou décapapétyl (si hommes ou pré-ménopausées).
- Vaccins vivants dans les 30 jours précédant la première dose de l’étude.
- Patients prenant actuellement les médicaments suivants, qui ne peuvent être interrompus 7 jours avant début du traitement :
- Tout médicament interdit selon la goséréline ou le décapapétyl (pré-ménopasique patients), l’étiquette du létrozole ou du ribociclib.
- Préparations/médicaments à base de plantes, compléments alimentaires.
- Médicaments qui ont un risque connu de prolonger l’intervalle QT ou de provoquer des torsades de Pointe.
- Médicaments à fenêtre thérapeutique étroite et majoritairement métabolisés via le CYP3A4.
- Inhibiteurs puissants du CYP3A4, y compris le pamplemousse, les hybrides de pamplemousse, les pomelos, caramboles et oranges de Séville.
- Inducteurs puissants du CYP3A4.
- Warfarine ou autre anticoagulant dérivé de la coumarine pour le traitement, la prophylaxie ou Par ailleurs. Le traitement par héparine, héparine de bas poids moléculaire ou fondaparinux est autorisé.
- Un WOCBP qui a un test de grossesse urinaire positif dans les 72 heures précédant l’attribution (voir Annexe 1). Si le test d’urine est positif ou ne peut être confirmé comme négatif, un un test de grossesse sérique sera nécessaire. Remarque : dans le cas où 72 heures se sont écoulées entre le test de grossesse de dépistage et la première dose du traitement à l’étude, un autre test de grossesse (urine ou sérum) doit être effectué et doit être négatif pour que le sujet commence à recevoir l’étude médicament.
- A des antécédents ou des preuves actuelles de toute condition, thérapie ou anomalie de laboratoire qui pourraient confondre les résultats de l’étude, interférer avec le sujet participation pendant toute la durée de l’étude, ou n’est pas dans le meilleur intérêt de le sujet à participer, de l’avis de l’investigateur traitant.
- A connu des troubles psychiatriques ou de toxicomanie qui interféreraient avec coopération avec les exigences de l’essai.
- Est enceinte ou allaite ou s’attend à concevoir ou engendrer des enfants dans la durée prévue de l’étude, en commençant par la visite de dépistage jusqu’à 120 jours après la dernière dose du traitement d’essai.
- Les hommes qui veulent engendrer des enfants devraient envisager de conserver le sperme avant de commencer le traitement par ribociclib.
- Personnes privées de liberté ou placées sous protection ou tutelle.
Calendrier prévisionnel
Lancement de l’étude : Mai 2022
Fin estimée des inclusions : Février 2030
Nombre de patients à inclure : 530
Coordonnateur de l'étude
Dr Paul COTTU – Institut Curie – CLCC Paris
Promoteur de l'étude
UNICANCER