IFCT-2403 Bi-MAPS
Evaluation de l’ivonescimab comme traitement de recours chez les patients atteints d’un mésothéliome pleural récidivant, précédemment traités par immunothérapie et chimiothérapie standard
Type d'essai : Académique / Institutionnel
Etat de l'essai : Ouvert
Situation thérapeutique : Tumeur solide ( Métastatique / Rechute )
Etablissement(s) participant(s)
Dr Clarisse AUDIGIER-VALETTE
Détails de l'essai
Objectif principal
Evaluer l’efficacité d’un anticorps bispécifique anti-VEGF/anti-PD-1 l’ivonescimab en 2ème ou 3ème ligne de traitement chez des patients présentant un mésothéliome pleural récidivant.
Objectif(s) secondaire(s)
Tolérance du traitement.
Activité du traitement.
Résumé / schéma de l'étude
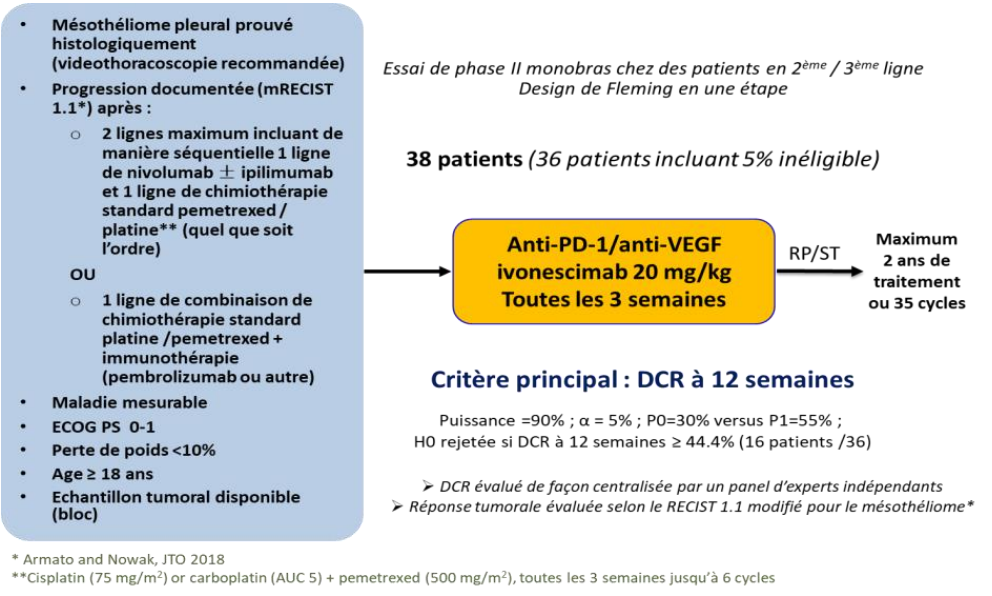
Critère(s) d'inclusion
- Consentement éclairé signé.
– Les patients doivent avoir signé et daté un formulaire de consentement éclairé écrit approuvé par un comité d’éthique conformément aux directives réglementaires et institutionnelles. Ce consentement doit être obtenu avant la réalisation de toute procédure liée au protocole qui ne fait pas partie de la prise en charge standard du patient.
– Les patients doivent être désireux et capables de se conformer aux visites prévues, au planning de traitement et aux tests de laboratoire. - Mésothéliome pleural histologiquement prouvé (pas de cytologie autorisée, biopsies par thoracoscopie recommandées).
Note : la certification de la pathologie par le réseau national d’experts NETMESO/MESOPATH doit avoir été faite comme cela est déjà fait en routine en France par l’expert régional NETMESO (ou une certification nationale similaire si le patient a obtenu son diagnostic de mésothéliome en dehors de France). - Progression documentée par tomodensitométrie avec injection d’iode selon RECIST 1.1 modifié pour le mésothéliome (mRECIST 1.1 ; épaississement pleural perpendiculaire à la paroi thoracique ou au médiastin de 7 mm ou plus, sur 2 positions, à trois niveaux distincts sur des coupes transversales de tomodensitométrie, séparées d’au moins 1 cm, la somme de 6 mesures définissant une mesure pleurale unidimensionnelle), ou selon RECIST 1.1 pour les ganglions médiastinaux ou les lésions métastatiques, après un maximum de 2 lignes incluant l’immunothérapie par nivolumab ± ipilimumab, et la chimiothérapie standard pemetrexed/platine [avec (40% du total des patients maximum) ou sans bevacizumab], séquentiellement (quel que soit l’ordre de traitement), ou une première ligne combinant chimiotherapie standard + immunothérapie (pembrolizumab ou autre médicament expérimental, mais pas de traitement anti-angiogénique).
- Maladie mesurable selon mRECIST 1.1.
- ECOG PS 0 ou 1.
- Perte de poids < 10% dans les 3 mois précédant l’entrée dans l’étude.
- Âge ≥ 18 ans.
- Espérance de vie > 3 mois.
- Échantillons pathologiques disponibles (au moins 10 lames des biopsies par thoracoscopie) pour des analyses centralisées de PD-L1, MTAP, immunohistochimie et NGS/ transcriptome, toutes les lames étant revues de manière centralisée par le panel français MESOPATH pour la certification histologique du mésothéliome. Si les tissus d’archives sont insuffisants ou indisponibles, le patient peut toujours être éligible après discussion avec l’IFCT.
- Fonctions biologiques adéquates : clairance de la créatinine ≥ 45 ml/min (Cockroft ou MDRD ou CKD-epi) ; neutrophiles≥ 1500/mm3; plaquettes ≥ 100 000/mm3; hémoglobine ≥ 9 g/dL ; ASAT et ALAT < 3 x LNS et bilirubine totale < 2 x LNS (les patients présentant des métastases hépatiques ou un syndrome de Gilbert doivent avoir des ASAT et ALAT ≤ 5 x LNS et une bilirubine totale de base ≤ 2 x LNS), protéines urinaires < 2+ ou quantification des protéines urinaires sur 24 heures < 1,0 g, taux de prothrombine (TP) ou (INR) ≤ 1,5 x LSN, et temps de prothrombine partielle (TTP) ou temps de thromboplastine partielle activée (TCA) ≤ 1,5 × LSN (sauf si les anomalies ne sont pas liées à une coagulopathie ou sont secondaires à une coagulation prophylactique).
- Les femmes en âge de procréer et sexuellement actives doivent utiliser une méthode de contraception hautement efficace dans les 28 jours précédant la première dose et pendant les 6 mois suivant la dernière dose de traitement. Les femmes doivent avoir un test de grossesse sérique ou urinaire négatif (sensibilité minimale de 25 UI/L ou unités équivalentes de HCG) dans les 24 heures précédant le début de la prise du médicament à l’étude.
- Pour les sujets masculins sexuellement actifs avec une partenaire en âge de procréer, une méthode de contraception efficace doit être utilisée pendant le traitement et pendant les 6 mois suivant la dernière dose.
- Patient bénéficiant d’une couverture par une assurance maladie nationale.
Critère(s) de non-inclusion
- ECOG PS > 1.
- Traitement antérieur du mésothéliome pleural par plus de 2 lignes de traitement systémique, et/ou par bevacizumab (ou un autre médicament anti-angiogénique/anti-VEGF), sauf s’il est associé à une chimiothérapie pemetrexed / platine (schéma validé par les lignes directrices de l’ASCO, du NCCN et de l’ESMO) avec un maximum de 40% du total des patients recrutés (n=15).
- Suspicion de maladie hyperprogressive ou de progression tumorale rapide lors d’un traitement par immunothérapie précédent (c’est-à-dire maladie progressive dans les 9 semaines suivant le traitement par nivolumab ± ipilimumab ou immunothérapie alternative, à partir du C1J1 ou de la randomisation en cas d’immunothérapie expérimentale précédente).
- Épanchement pleural comme seule anomalie radiologique sans épaississement pleural mesurable ni hypertrophie des ganglions médiastinaux.
- Mésothéliome péritonéal, péricardique ou de la tunique vaginale du testicule, sans atteinte pleurale au moment du diagnostic.
- Diagnostic antérieur d’adénocarcinome de n’importe quel site anatomique au cours des 3 années précédentes, à l’exception d’antécédents d’adénocarcinome de la prostate ; si cancer de la prostate localisé, avec des facteurs de bon pronostic selon la classification de d’Amico (< T2a, score de Gleason ≤ 6 et PSA ≤ 10 ng/ml), et à condition qu’il ait été traité selon une modalité curative (chirurgie ou radiothérapie, en l’absence de toute chimiothérapie).
- Antécédent de cancer au cours des 3 dernières années ou cancer actif (à l’exception d’un carcinome in situ du col de l’utérus déjà traité, ou d’un cancer basocellulaire de la peau, traité ou non).
- Épanchement pleural non contrôlé malgré une pleurodèse antérieure (talc ou autre), nécessitant une thoracocentèse (par aspiration pleurale) plus souvent que tous les 21 jours. Toutefois, les patients ayant un cathéter pleural à demeure efficace sont éligibles.
- Métastases cérébrales symptomatiques non traitées (sans radiothérapie cérébrale in toto ou radiothérapie cérébrale ablative stéréotaxique préalable ou sans résection chirurgicale). Un délai d’au moins 2 semaines entre la fin de la radiothérapie et le début du traitement doit être respecté. Les métastases cérébrales asymptomatiques ne nécessitant pas de corticothérapie à une dose >10 mg de prednisone ou équivalent par jour ou de perfusions de mannitol sont autorisées.
- Radiothérapie nécessaire au début du traitement, à l’exception de la radiothérapie palliative osseuse sur un site tumoral douloureux ou compressif (dans ce cas, les lésions cibles ne doivent pas être dans la zone irradiée).
- Antécédents d’immunodéficience primaire, de transplantation d’organe nécessitant un traitement immunosuppresseur, de prise de tout médicament immunosuppresseur dans les 28 jours précédant la date d’inclusion, ou antécédents de toxicité sévère (grade 3/4) par mécanisme immunitaire liée à un autre traitement d’immunothérapie pour n’importe quel type de maladie.
- Traitement systémique par corticothérapie à une dose >10 mg de prednisone ou équivalent par jour, dans les 14 jours précédant le début du traitement. Les corticostéroïdes inhalés, nasaux ou thématiques sont autorisés.
- Antécédents de maladie auto-immune active nécessitant un traitement immunosuppresseur systémique, y compris, mais sans s’y limiter, polyarthrite rhumatoïde, myasthénie, hépatite auto-immune, lupus systémique, granulomatose de Wegener, thrombose vasculaire associée au syndrome des anti-phospholipides, syndrome de Sjögren avec maladie pulmonaire interstitielle, syndrome de GuillainBarré au cours des 15 dernières années, sclérose en plaques, vascularite ou glomérulonéphrite.
Les patients atteints de diabète de type I, d’hypothyroïdie, d’une maladie cutanée immunitaire (vitiligo, psoriasis, alopécie) ou d’une polyarthrite rhumatoïde bénigne ne nécessitant pas de traitement systémique immunosuppresseur ou plus de 10 mg par jour de corticoïdes oraux, ou syndrome de sicca bénin (Sjogren) sans maladie pulmonaire interstitielle, ou antécédents de syndrome de Guillain-Barre depuis plus de 15 ans, totalement réversible et sans séquelles, sans traitement immunosuppresseur systémique au cours des 20 dernières années, sont autorisés à être inclus. Les patients atteints de la maladie de Grave et/ou de psoriasis ne nécessitant pas de traitement systémique au cours des deux dernières années suivant l’inclusion ne sont pas exclus. - Maladie intestinale inflammatoire active (diverticulose, maladie de Crohn, recto-colite hémorragique, maladie cœliaque) ou toute maladie intestinale chronique grave avec diarrhée incontrôlée.
- Maladie interstitielle pulmonaire préexistante, modérée ou grave, évaluée par le scanner diagnostique et diminution de la TLCO de plus de 35 % par rapport aux valeurs normales théoriques liées à une telle maladie interstitielle (PID).
- Interventions chirurgicales majeures ou traumatismes graves dans les 4 semaines précédant l’inclusion ou projets d’interventions chirurgicales majeures dans les 4 semaines suivant la première dose (selon la décision de l’investigateur). Interventions locales mineures (à l’exclusion du cathétérisme veineux central et de l’implantation d’un port) dans les 3 jours précédant l’inclusion.
- Antécédents de tendances hémorragiques ou de coagulopathie et/ou symptômes ou risques hémorragiques cliniquement significatifs dans les 4 semaines précédant l’inclusion, y compris, mais sans s’y limiter : Saignement gastro-intestinal ; hémoptysie (définie comme l’expectoration de ≥ 0,5 cuillère à café de sang frais ou de petits caillots sanguins). Note : l’hémoptysie transitoire associée à une bronchoscopie diagnostique est autorisée ; saignement nasal/épistaxis (un écoulement nasal sanglant est autorisé) ; nécessité d’un traitement anticoagulant thérapeutique dans les 14 jours précédant l’inclusion. Note : l’anticoagulation prophylactique pour une thrombose veineuse profonde/embolie pulmonaire ou pour maintenir la perméabilité veineuse est autorisée.
- Hypertension actuelle avec une pression artérielle systolique ≥ 150 mmHg ou une pression artérielle diastolique ≥ 100 mmHg après un traitement antihypertenseur oral.
- Antécédents de maladies majeures avant l’inclusion, en particulier :
- Angor instable, infarctus du myocarde, insuffisance cardiaque congestive (classification de la New York Heart Association [NYHA] ≥ grade 2) ou maladie vasculaire (par exemple, anévrisme aortique avec risque de rupture) ayant nécessité une hospitalisation dans les 12 mois précédant l’inclusion, ou autre déficience cardiaque susceptible d’affecter l’évaluation de la sécurité du médicament à l’étude (par exemple, arythmie mal contrôlée, ischémie myocardique). Les patients ayant des antécédents cardiaques significatifs, même s’ils sont contrôlés, doivent avoir une FEVG > 45 %.
- Antécédents de varices œsophagiennes et gastriques, d’ulcères graves, de plaies qui ne guérissent pas, de fistules abdominales, d’abcès intra-abdominaux ou d’hémorragies gastro-intestinales aiguës dans les 6 mois précédant l’inclusion.
- Antécédents d’accident thromboembolique artériel, d’accident thromboembolique veineux de grade 3 ou plus tel que spécifié par le National Cancer Institute (NCI) Common Terminology Criteria for Adverse Events (CTCAE) 5.0, d’accident ischémique transitoire, d’accident vasculaire cérébral, de crise hypertensive ou d’encéphalopathie hypertensive dans les 6 mois précédant l’inclusion.
- Exacerbation aiguë d’une maladie pulmonaire obstructive chronique dans les 4 semaines précédant l’inclusion.
- Antécédents de perforation du tractus gastro-intestinal et/ou de fistule, antécédents d’obstruction gastro-intestinale (y compris obstruction intestinale incomplète nécessitant une alimentation parentérale), résection intestinale étendue (colectomie partielle ou résection étendue de l’intestin grêle) dans les 6 mois précédant l’inclusion.
- Imagerie réalisée au cours de la période de screening montrant que le patient présente :
- Preuve radiologiquement documentée d’une invasion des vaisseaux sanguins pulmonaires majeurs ou d’un encapsulage par le cancer.
- Des signes radiographiques de cavitation pulmonaire intra tumorale.
- Infection active non contrôlée, y compris tuberculose active, hépatite virale aiguë connue B et C selon les tests sérologiques. Les patients présentant des séquelles sérologiques d’une hépatite virale guérie peuvent être inclus. Les antécédents de tuberculose pulmonaire primaire chez les jeunes ne constituent pas une contreindication. Les antécédents de tuberculose ne constituent pas une contre-indication si le patient a été traité pendant au moins 6 mois par un traitement antibiotique antituberculeux.
- Antécédents connus de test positif au VIH ou au SIDA, sans avoir reçu de thérapie antirétrovirale efficace au cours des 4 dernières semaines et présentant une charge virale VIH > 200 copies/ml, indépendamment du nombre de lymphocytes T CD4+.
- Vaccin vivant atténué reçu dans les 30 jours précédents ; les vaccins anti-SARS-CoV2 à vecteur ARNm et adénovirus sont autorisés.
- Incapacité à se conformer à l’étude et/ou aux procédures de suivi, telle qu’estimée par l’investigateur référent.
- Allergie ou hypersensibilité connue au traitement de l’étude ou à tout excipient.
- Traitement concomitant avec un autre traitement expérimental ou participation à un autre essai clinique.
- Patient faisant l’objet d’une protection juridique ou incapable d’exprimer sa volonté.
Calendrier prévisionnel
Lancement de l’étude : Avril 2025
Fin estimée des inclusions : Mars 2028
Nombre de patients à inclure : 38
Information(s) complémentaire(s)
Prélèvements tissulaires
Les biopsies réalisées au moment du diagnostic seront collectées.
Collecte de tissus congelés à partir de biopsies chirurgicales par thoracoscopie (au moment du diagnostic initial du mésothéliome pleural, ou de biopsies avant l’inclusion si les patients sont rebiopsiés).
De même, de nouvelles biopsies tumorales au moment de la progression avec le médicament à l’étude sont encouragées (si elles sont nécessaires pour des raisons médicales) et utilisées pour la même analyse ancillaire.
Prélèvements sanguins
Des échantillons sanguins seront prélevés à l’inclusion, à la 1ère évaluation (12 semaines) et à la progression. Un tube cell-Free DNA de 10mL et 2 tubes EDTA de 10mL seront prélevé à chaque point de prélèvement.
Coordonnateur de l'étude
Pr Arnaud SCHERPEREEL – CHU Lille
Promoteur de l'étude
Intergroupe Francophone de Cancérologie Thoracique (IFCT)