Prodige 73 - LOGICAN
Etude de phase II randomisée évaluant la combinaison trifluridine/tipiracil plus oxaliplatine versus FOLFOX chez des patients inéligibles à une trichimiothérapie porteurs d’un adénocarcinome gastrique, de l’œsophage ou de la jonction œsogastrique localement avancé, en rechute ou métastatique
Phase : II
Type d'essai : Académique / Institutionnel
Etat de l'essai : Ouvert
Situation thérapeutique : Tumeur solide ( Métastatique / Rechute , Localement avancée / Non résécable )
Type d'essai : Académique / Institutionnel
Etat de l'essai : Ouvert
Situation thérapeutique : Tumeur solide ( Métastatique / Rechute , Localement avancée / Non résécable )
Etablissement(s) participant(s)
Détails de l'essai
Objectif principal
L’objectif principal de cette étude est d’évaluer la supériorité en termes de survie sans progression (SSP) de l’association trifluridine/tipiracil + oxaliplatine sur le traitement FOLFOX.
Résumé / schéma de l'étude
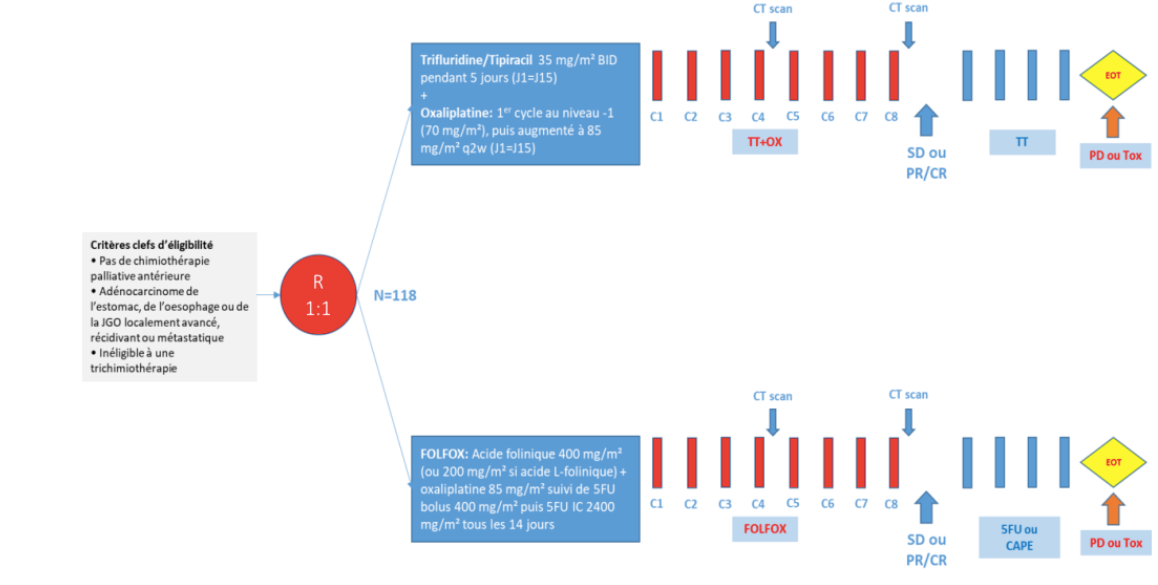
Critère(s) d'inclusion
- Adénocarcinome non résécable de l’estomac, de l’œsophage ou de la jonction gastro-oesophagienne (JGO) localement avancé, récurrent ou métastatique, confirmé par histologie, inéligible à un traitement curatif et inéligible à un traitement par immunothérapie (CPS < 5 ou patient présentant des contre indications à l’utilisation d’un traitement par immunothérapie).
- Absence de dysphagie ou de difficulté à déglutir.
- Pas de surexpression/amplification d’HER2 (IHC 0 ou 1+; si IHC 2+, l’HIS doit être négative). Score CPS PD-L1 connu (résultats en % avec le nom de la méthode utilisée). Les statuts microsatellites et MMR de la tumeur du patient (MSI/MSS, pMMR/dMMR) doivent également être connus au moment de la sélection du patient.
- Au moins une lésion évaluable selon les critères RECIST 1.1 en dehors de toute zone précédemment irradiée.
- Pas de chimiothérapie palliative antérieure.
- Age ≥ 18 ans.
- Patient non éligible pour un triplet de chimiothérapie = ECOG-PS = 2 ou âgés de ≥ 70 ans + un critère de fragilité au score ADL ou IADL ou dénutrition définie par un taux d’albumine < 30 g/l). La raison d’inéligibilité à la trichimiothérapie sera collectée dans le CRF.
- Fonction des organes suffisantes :
- Neutrophiles ≥ 1.5×109/L.
- Plaquettes ≥ 100×109/L.
- Hémoglobine ≥ 9 g/L.
- Bilirubine sérique < 2 x LSN , jusqu’à 2.5 x LSN en cas de métastases hépatiques (drainage biliaire autorisé).
- Transaminases < 5 x LSN.
- Clairance de la créatinine > 40 mL/min.
- Absence de déficience en dihydropyrimidine déhydrogénase (DPD) (uracilémie < 16 ng/ml).
- Les femmes en âge de procréer doivent avoir un test de grossesse sérique ou urinaire négatif dans les 14 jours qui précèdent le début du traitement de l’étude.
- Les patients doivent accepter d’utiliser des méthodes de contraception adéquates pendant la durée du traitement à l’étude et jusque 6 mois après l’arrêt du traitement à l’étude.
- Les patients doivent être affiliés au système de la Sécurité Sociale (ou à un système équivalent).
- Le/la patient(e) doit avoir signé et daté un formulaire de consentement éclairé écrit avant toute procédure spécifique à l’essai. Si le patient est physiquement incapable de donner son consentement écrit, une personne de confiance de leur choix, indépendante de l’investigateur ou du promoteur, peut confirmer par écrit le consentement du patient.
- Disponibilité de matériel tumoral archivé pour les études ancillaires.
Critère(s) de non-inclusion
- Patient avec un statut de performance ECOG PS > 2.
- Autre tumeur maligne présente ou antérieure au cours des 3 années précédentes (sauf carcinome à cellules squameuses cutané traité par chirurgie).
- Chimiothérapie adjuvante ou radiochimiothérapie terminée depuis moins de 6 mois.
- Neuropathie périphérique de grade NCI ≥ 2 à la baseline.
- Patients avec allergie connue ou hypersensibilité sévère à d’un des médicaments à l’essai ou à l’un des excipients des médicaments à l’essai.
- Patients incapables de se conformer aux obligations de l’essai pour des raisons géographiques, sociales ou physiques, ou incapables de comprendre l’objectif et les procédures de l’étude.
- Traitement antérieur par trifluridine/tipiracil.
- Infection connue par le virus de l’immunodéficience humaine (VIH).
- Infection active par le virus de l’hépatite B (VHB, définie par un test positif de détection de l’antigène de surface du virus de l’hépatite B [HBsAg] avant l’inclusion) ou le virus de l’hépatite C (VHC).
- Pneumopathie interstitielle.
- Antécédent de pneumonie ayant nécessité un traitement par corticostéroïde systémique.
- Infections actives.
- Patiente enceinte ou allaitante.
- Participation à un autre essai clinique dans les 30 jours qui précèdent la randomisation.
- Personne privé de leur liberté, sous protection juridique ou sous tutelle.
- Maladie des artères coronaires significative ou histoire d’infarctus du myocarde dans les 12 derniers mois, ou haut risque d’arythmie non contrôlée (pour les hommes : QTc ≥ 450 msec, pour les femmes: QTc ≥ 470 msec).
Calendrier prévisionnel
Lancement de l’étude : Décembre 2022
Fin estimée des inclusions : Janvier 2026
Nombre de patients à inclure : 118
Coordonnateur de l'étude
Dr Christelle DE LA FOUCHARDIERE
Centre Léon Bérard – CLCC Lyon
Promoteur de l'étude
UNICANCER
Dernière mise à jour le 15 juillet 2024