Prodige 69 - FOLFIRINEC
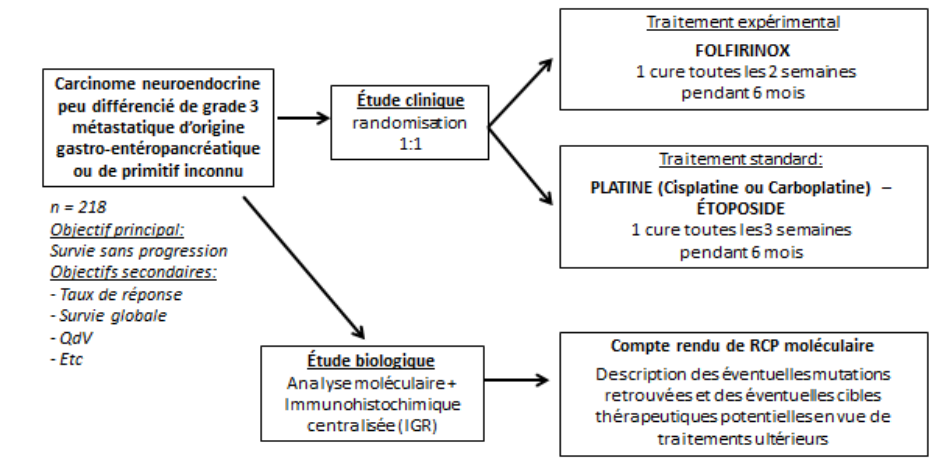
Etude de phase II évaluant l’efficacité du mFOLFIRINOX vs platine – etoposide chez les patients atteints de carcinomes neuro-endocrines peu différenciés de grade 3 métastatiques gastro-entero-pancréatiques et de primitif inconnu avec l’établissement d’un profil moléculaire à la recherche de cibles thérapeutiques
Type d'essai : Académique / Institutionnel
Etat de l'essai : Ouvert
Situation thérapeutique : Tumeur solide ( Métastatique / Rechute )
Etablissement(s) participant(s)
Dr Claire JARAUDIAS
Dr Yves RINALDI
Dr Laetitia DAHAN
Dr Thibault BROTELLE
Dr Laurent MINEUR
Détails de l'essai
Objectif principal
Comparer la survie sans progression (SSP) entre mFOLFIRINOX et platine – étoposide évaluée par l’investigateur selon les critères RECIST 1.1.
Objectif(s) secondaire(s)
SSP selon la relecture centralisée (critères RECIST v1.1).
Taux de meilleure réponse objective (TRO).
Survie globale (SG).
Evènements indésirables selon le NCI CTC V4.0.
Dose intensité – réductions de dose.
Qualité de vie évaluée par les questionnaires EORTC, QLQ-C30 et EQ-5D-5L.
Etablir un profil moléculaire dans les 2 mois après l’envoi de l’échantillon de la tumeur pour chaque patient inclus dans l’étude et fournir un compte-rendu de RCP moléculaire à l’investigateur prenant en charge le patient.
Pourcentage de perte d’expression de Rb (indépendamment du sous-groupe petites cellules ou grandes cellules).
Corrélation entre TRO, SSP et SG et les altérations moléculaires (Rb, TP53, MSH2…) dans les deux bras de traitement.
Résumé / schéma de l'étude
Critère(s) d'inclusion
- Carcinome neuroendocrine peu différencié de grade 3 ou MINEN avec composante de carcinome neuroendocrine peu différencié de grade 3 représentant ≥ 30% de la tumeur.
- Tumeur primitive d’origine gastro-entéro-pancréatique ou inconnue.
- Sous types histologiques incluables : à petites cellules ou à grandes cellules ou non à petites cellules ou non typable.
- Maladie métastatique.
- 1ère ligne de traitement pour une maladie métastatique. Pas de chimiothérapie ou de traitement systémique à visée antitumorale préalable.
- Au moins une lésion mesurable selon les critères RECIST 1.1 évaluée par scanner ou IRM.
- Bloc tumoral disponible.
- PNN≥ 1.5 x 109/l, plaquettes≥ 100 x 109/l et hémoglobine > 8 g/dl.
- Bilirubine totale ≤ 1.5 x LSN, ASAT ≤ 2.5N, ALAT≤ 2.5N or ASAT/ALAT ≤ 5 x LSN en cas de métastases hépatiques.
- Age ≥ 18 ans.
- Status de performance ECOG ≤ 1.
- Patient en capacité de comprendre les modalités de l’essai thérapeutique, de signer le consentement éclairé et de se conformer aux exigences du protocole.
- Les femmes en âge de procréer ainsi que les hommes (ayant des rapports sexuels avec des femmes en âge de procréer) doivent s’engager à utiliser des moyens de contraception efficaces tout au long de l’étude et au cours des 6 mois suivant l’administration de la dernière dose du médicament à l’étude.
- Patient affilié à la sécurité sociale.
Critère(s) de non-inclusion
- Tumeur neuroendocrine de grade 3 bien différenciée selon la classification OMS 2017.
- Insuffisance rénale sévère (clairance de la créatinine inférieure à 30mL/min, MDRD).
- Déficit partiel ou complet en Dihydropyrimidine Deshydrogenase (DPD) (uracilémie ≥ 16 ng/mL).
- Maladie de Gilbert.
- Neuropathie pré-éxistante permanente (grade ≥ 2 NCI CTC V4.0).
- Traitement préalable par chimiothérapie ou thérapie ciblée.
- Métastases cérébrales sauf si asymptomatiques ou stables sous corticothérapie pendant deux semaines. La radiothérapie est nécessaire avant inclusion en cas de symptômes liés à ces métastases cérébrales.
- Traitement par sorivudine et autres analogues inhibiteurs de la DPD comme la brivudine.
- Traitement par millepertuis (Hypericum perforatum).
- Femme enceinte ou allaitante.
- Patient séropositif pour VIH, hépatite B ou C ou autres syndromes d’immunodéficience.
- Antécédent de pathologie maligne dans les trois dernières années à l’exception du carcinome basocellulaire de la peau ou du carcinome in situ du col utérin correctement traités.
- Maladie chronique non contrôlée ou infection active incompatible avec une participation à l’étude.
- Vaccination (vaccin vivant) dans les 30 jours précédant le début du traitement.
- Personne privée de liberté ou sous tutelle.
- Intervalle QT/QTc > 450 msec pour les hommes et > 470 msec pour les femmes à l’ECG.
- K+ < LIN, Mg²+ < LIN, Ca²+ < LIN.
- Antécédents ou hypersensibilité connue à l’un des agents de chimiothérapie de l’étude ou à leurs excipients.
- Patient participant à un autre essai clinique en cours de traitement ou dont le traitement s’est terminé moins de quatre semaines avant l’inclusion.
Calendrier prévisionnel
Lancement de l’étude : Octobre 2020
Fin estimée des inclusions : Septembre 2027
Nombre de patients à inclure : 218
Information(s) complémentaire(s)
Étude ancillaire :
L’étude FOLFIRINEC est couplée à la réalisation d’un profil moléculaire de chaque tumeur par NGS, et immunohistochimie. Ce profil moléculaire fera l’objet d’une discussion en RCP moléculaire. Le compte-rendu de RCP moléculaire sera envoyé à l’investigateur durant l’essai clinique environ deux mois après réception du bloc tumoral. Ce profil moléculaire permettra aussi d’explorer les biomarqueurs de réponse à la chimiothérapie. Des échantillons sanguins seront également collectés pour l’analyse de l’AND tumoral circulant.
Coordonnateur de l'étude
Dr Julien HADOUX
Institut Gustave Roussy
Promoteur de l'étude
CHU DIJON