ATOMYELO
Etude de phase I d’escalade de dose et d’expansion évaluant la tolérance et l’efficacité de l’Arsenic oral (ATO) dans les Syndromes myélodysplasiques de faible risque en échec aux Agents Stimulants l’Erythropoïèse et au Luspatercept (ou non éligibles à ce dernier)
Type d'essai : Académique / Institutionnel
Etat de l'essai : Ouvert
Situation thérapeutique : Hématologie
Etablissement(s) participant(s)
Pr Thomas CLUZEAU
Détails de l'essai
Objectif principal
Partie I (Etude de Phase I) :
Déterminer la dose limitante de toxicité de l’ATO oral.
Partie II (Phase d’expansion) :
Déterminer le taux de réponse érythroïde (HI-E) sous ATO oral sur 12 semaines.
Objectif(s) secondaire(s)
Déterminer la tolérance et le profil de toxicité mesurés selon les CTCAE.
Pharmacocinétique : PK/PD.
Déterminer le taux de réponse (RC + RP + maladie stable avec amélioration hématologique selon les critères IWG 2018).
Déterminer la durée de la réponse jusqu’à la progression et la perte de l’indépendance transfusionnelle chez les patients répondeurs.
Déterminer le taux et le délai de transformation en SMD de haut risque ou LAM.
Déterminer la survie sans progression.
Déterminer la survie globale.
Analyses exploratoires : Identifier les facteurs associés à la survie et à la réponse, incluant l’IPSS-R, le caryotype et les mutations somatiques (IPSSM).
Résumé / schéma de l'étude
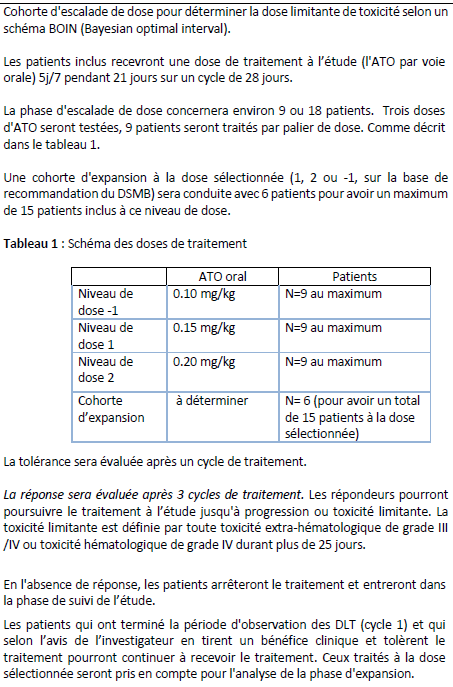
Critère(s) d'inclusion
- Syndrome myélodysplasique selon la classification OMS 2022.
- Âge ≥ 18 ans.
- Patient avec un SMD de faible risque selon la classification IPSS-R (très faible, faible, intermédiaire) :
- Non sidéroblastique en échec d’ASE ou en rechute après ASE (au moins 12 semaines d’Epoïétine alfa à 60000UI ou équivalent) sans progression de la maladie ou inéligible aux ASE (défini par un taux d’EPO>500UI/L).
- Sidéroblastique en échec d’ASE ou en rechute après ASE (au moins 12 semaines d’Epoïétine alfa à 60000UI ou équivalent) ou inéligible aux ASE (défini par un taux d’EPO>500UI/L) et en échec ou en rechute après Luspatercept.
- Del (5q) en échec d’ASE ou en rechute après ASE (au moins 12 semaines d’Epoïétine alfa à 60000UI ou équivalent) et en échec ou en rechute après Lenalidomide.
- Dépendance transfusionnelle (au moins 3 CG nécessaires dans une période de 16 semaines et au moins 2 épisodes de transfusions durant cette période).
- Patient non éligible à un autre essai clinique.
- Fonction rénale adéquate définie par un taux de créatinine inférieur à 1.5 x LSN et clairance de la créatinine ≥ 40mL/min (selon formule MDRD).
- Fonction hépatique adéquate définie par un taux de bilirubine totale et de transaminases inférieurs à 1.5 x LSN.
- Patient non réfractaire aux transfusions de plaquettes.
- Consentement écrit.
- Le patient doit comprendre et signer volontairement le consentement éclairé.
- Le patient doit être capable de respecter le calendrier des visites protocolaires prévues dans le cadre de l’étude et suivre les exigences du protocole.
- ECOG ≤ 2 au moment du screening.
- Les patients diabétiques doivent être équilibrés avant l’inclusion avec un taux d’HbA1C ≤ 7.5%.
- Pour cette étude, une femme en âge de procréer est définie comme une femme sexuellement mature qui : (1) n’a pas subi d’hystérectomie et d’ovariectomie bilatérale ; ou (2) n’est pas naturellement ménopausée (une aménorrhée consécutive à un traitement anti-cancéreux n’exclut pas la possibilité de procréer) depuis au moins 24 mois consécutifs (c’est-àdire qu’elle a eu des menstruations à un moment quelconque au cours des 24 précédents mois). Une femme en âge de procréer participant à l’étude doit :
- Avoir eu 2 tests de grossesse négatifs avant le début du traitement protocolaire (sauf si le test de grossesse a été fait dans les 72h avant le jour 1 du cycle 1). Elle doit accepter de réaliser des tests de grossesse mensuels durant l’étude et après la sortie d’essai.
- Si la patiente est sexuellement active, elle doit accepter d’utiliser et de respecter une contraception hautement efficace** sans interruption, 5 semaines avant le début du traitement protocolaire, pendant le traitement protocolaire (y compris pendant les interruptions de traitement) et pendant 24 semaines après l’arrêt du traitement protocolaire.
- ** Dans le cadre du protocole, une contraception hautement efficace est définie comme suit (information figurant également dans le consentement) : contraception hormonale (par exemple : pilule contraceptive, injection, implant, patch transdermique, anneau vaginal), dispositif intra-utérin, ligature des trompes ou partenaire ayant subi une vasectomie.
- Les hommes doivent accepter d’utiliser un préservatif, défini comme un préservatif masculin en latex ou un préservatif masculin sans latex NON fabriqué à partir d’une membrane naturelle (animale) (par exemple, le polyuréthane), lors de chaque rapport sexuel avec une femme enceinte ou en âge de procréer pendant toute la durée du traitement, pendant les interruptions de traitement et pendant au moins 24 semaines après l’arrêt du traitement, même s’il a subi une vasectomie.
Critère(s) de non-inclusion
- Infection sévère ou toute autre affection sévère non contrôlée.
- Hypertension non contrôlée.
- Maladie cardiaque significative – Classe NYHA III ou IV ou infarctus du myocarde au cours des 6 derniers mois.
- QTcF > 460ms.
- Utilisation d’agents en cours d’investigation dans les 30 jours ou de tout agent anticancéreux (incluant les IMID) dans les 2 semaines précédant l’inclusion, à l’exception de l’hydroxyurée. Le patient doit avoir récupéré (grade≤1) de toutes les toxicités induites par un traitement préalable. Par contre, les patients peuvent avoir reçu du Lenalidomide, un hypométhylant, ou du sérum anti lymphocytaire (SAL) (mais pas dans les 4semaines précédentes et, pour le SAL, les 16 semaines précédentes).
- Utilisation d’EPO dans les 4 semaines précédant l’inclusion.
- Cancer actif ou diagnostic de cancer au cours de l’année précédant l’entrée dans l’essai, exceptés les carcinomes basocellulaires et les
carcinomes in situ du col de l’utérus ou du sein. - Patient déjà inclus dans un essai thérapeutique avec une molécule expérimentale.
- Infection connue par le VIH ou hépatite B ou hépatite C active.
- Femmes enceintes ou allaitantes.
- Toute affection médicale ou psychiatrique ne permettant pas au patient de comprendre ou de signer le consentement éclairé.
- Patient éligible à une allogreffe de cellules souches hématopoïétiques.
- Patients présentant une hypoxie nécessitant une assistance en oxygène.
- Patient présentant un risque excessif d’encéphalopathie (c’est-à-dire une carence en vitamine B1).
- Patient recevant un traitement connu pour allonger l’intervalle QT.
- Patient présentant une hypersensibilité connue à l’arsenic ou à l’un des excipients.
- Personnes non affiliées à un régime de sécurité sociale ou équivalent.
- Personnes privées de liberté par décision judiciaire ou administrative.
- Personnes faisant l’objet d’une mesure de protection légale (tutelle, curatelle, sauvegarde de justice).
Calendrier prévisionnel
Lancement de l’étude : Juillet 2025
Fin estimée des inclusions : Juillet 2026
Nombre de patients à inclure : 24
Coordonnateur de l'étude
Pr Thomas CLUZEAU – CHU de Nice
Promoteur de l'étude
Groupe Francophone des Myélodysplasies (GFM)