ISIDE
Essai ouvert multicentrique et à bras unique de phase IIIB visant à évaluer la sécurité et l’efficacité du sacituzumab govitécan chez les patients présentant un cancer du sein triple négatif métastatique avec une analyse des biomarqueurs
Phase : III
Type d'essai : Académique / Institutionnel
Etat de l'essai : Ouvert
Situation thérapeutique : Tumeur solide ( Métastatique / Rechute , Localement avancée / Non résécable )
Type d'essai : Académique / Institutionnel
Etat de l'essai : Ouvert
Situation thérapeutique : Tumeur solide ( Métastatique / Rechute , Localement avancée / Non résécable )
Etablissement(s) participant(s)
Pr Anthony GONCALVES
Détails de l'essai
Objectif principal
L’objectif principal est d’évaluer l’efficacité du sacituzumab govitecan via le taux de réponse objective (ORR) évalué par l’investigateur selon RECIST v1.1.
Objectif(s) secondaire(s)
Survie sans progression (PFS).
Durée de la réponse (DOR).
Risque de bénéfice clinique (CBR).
Survie globale (OS).
L’incidence des événements indésirables (sécurité).
Résumé / schéma de l'étude
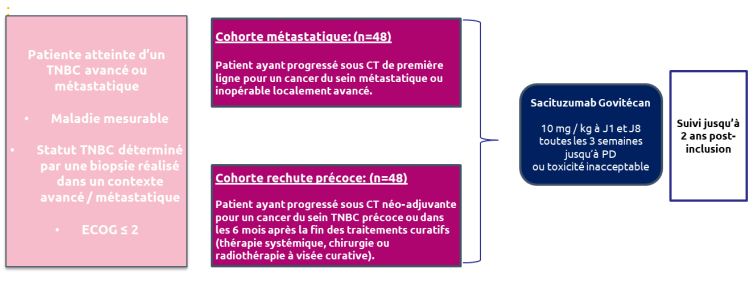
Critère(s) d'inclusion
- Le patient doit avoir signé un consentement éclairé écrit avant toute procédure spécifique à l’essai.
- Homme ou femme ≥ 18 ans.
- Patientes atteintes d’un cancer du sein triple négatif (mTNBC) localement avancé, inopérable ou métastatique, documenté pathologiquement, dont la maladie a progressé soit avec une chimiothérapie de 1ère ligne avec ou sans inhibiteurs de point de contrôle immunitaire (ICI) soit avec un traitement ciblé (par ex. inhibiteur de l’AKT, inhibiteur de la PI3K, inhibiteur de la PARP) pour le cancer du sein métastatique ou inopérable localement avancé (ABC) ou sous chimiothérapie (néo)adjuvante avec ou sans immunothérapie pour le TNBC précoce ou dans les 6 mois après la fin de toute thérapie systémique, chirurgie ou radiothérapie avec une intention curative, quoi qu’il arrive en dernier. Le nombre de patients en progression précoce sera plafonné à 20 % de la population globale.
- Exposition antérieure à un taxane dans un contexte localisé ou avancé/métastatique.
- Maladie mesurable, telle que définie par RECIST v1.1.
- Le patient doit avoir accepté d’effectuer des biopsies avant, pendant et après le traitement. Si le médecin envisage de faire la biopsie sur le site tumoral primitif pour des raisons d’accessibilité, elle ne peut être réalisée que si le site tumoral primitif n’a pas été préalablement irradié.
- Avoir un site métastatique facilement accessible à la biopsie (à l’exception des métastases osseuses).
- Statut de performance de l’Eastern Cooperative Oncology Group (ECOG) ≤ 2.
- Espérance de vie ≥ 12 semaines.
- Fonction hématologique et organique adéquate.
- Test négatif de l’antigène de surface de l’hépatite B (HBsAg) lors du dépistage (les patients avec un test HBsAg négatif et un test positif des anticorps anti-hépatite B totaux (HBcAb) lors du dépistage sont éligibles), test négatif des anticorps contre le virus de l’hépatite C (VHC) lors du dépistage, ou positif. Test d’anticorps du VHC suivi d’un test d’ARN du VHC négatif lors du dépistage.
- Preuve du statut post-ménopausique ou test urinaire de grossesse négatif dans les 72 heures ou test de grossesse sérique dans les 14 jours suivant le traitement à l’étude et confirmé avant le traitement au cycle 1 jour 1 (C1D1) pour les patientes préménopausées.
- La femme en âge de procréer et le patient de sexe masculin doivent accepter d’utiliser une contraception adéquate pendant toute la durée de la participation à l’essai et jusqu’à 6 mois après la fin du traitement pour les femmes et jusqu’à 3 mois pour les hommes.
- Patient affilié à un système de sécurité sociale (ou équivalent).
- Le patient est disposé et capable de se conformer au protocole pendant toute la durée de l’essai, y compris le traitement et les visites programmées, et les examens, y compris le suivi.
Critère(s) de non-inclusion
- Participation à un autre essai thérapeutique dans les 30 jours précédant l’inscription.
- Métastases du système nerveux central (SNC) symptomatiques, non traitées ou en progression active ou signes de maladie leptoméningée ou de compression de la moelle épinière cliniquement active. Les patients présentant des métastases cérébrales stables et asymptomatiques seront éligibles, mais le nombre sera plafonné à 15 % de la population globale.
- Antécédents de cancer autre que mTNBC dans les 5 ans précédant C1D1, à l’exception de ceux présentant un risque négligeable de métastases ou de décès (par exemple, taux de SG sur 5 ans > 90 %) et traités avec une intention curative (par ex. carcinome in situ du col de l’utérus, carcinome cutané non mélanique, cancer localisé de la prostate, carcinome canalaire in situ ou cancer de l’utérus de stade I).
- Satisfait à l’un des critères suivants de maladie cardiaque :
- Infarctus du myocarde ou angine de poitrine instable dans les 6 mois suivant l’inscription.
- Antécédents d’arythmie ventriculaire grave (c’est-à-dire de tachycardie ventriculaire ou de fibrillation ventriculaire), de bloc auriculo-ventriculaire de haut grade ou d’autres arythmies cardiaques nécessitant des médicaments antiarythmiques (à l’exception de la fibrillation auriculaire bien contrôlée par des médicaments antiarythmiques) ; antécédents d’allongement de l’intervalle QT.
- Insuffisance cardiaque congestive de classe III ou supérieure de la New York Heart Association (NYHA) ou fraction d’éjection ventriculaire gauche < 40 %.
- Infection sévère non contrôlée nécessitant des antibiotiques oraux ou IV dans les 4 semaines précédant C1D1.
- Intervention chirurgicale majeure dans les 4 semaines précédant C1D1.
- Antécédents de réactions allergiques, anaphylactiques ou autres réactions d’hypersensibilité graves aux anticorps humanisés.
- Hypersensibilité connue au médicament à l’étude, à ses métabolites ou à l’excipient de la formulation.
- Patients recevant des traitements anticancéreux concomitants tels que la chimiothérapie, l’immunothérapie, l’hormonothérapie et la radiothérapie.
- Patients présentant des toxicités non résolues d’un traitement anticancéreux antérieur, définies comme des toxicités (autres que l’alopécie) non encore résolues selon les critères de terminologie communs pour les événements indésirables du National Cancer Institute (NCI-CTCAE) v5.0 gradev> 2.
- Traitement avec des corticostéroïdes systémiques dosés à> 20 mg de prednisone ou équivalent ou d’autres médicaments immunosuppresseurs systémiques dans les 2 semaines précédant C1D1.
- Antécédents connus de test positif pour le VIH ou syndrome d’immunodéficience acquise connu.
- Infection au Covid-19 lors du dépistage.
- Preuve d’une maladie concomitante significative non contrôlée.
- Les personnes ayant des conditions physiques ou psychologiques considérées comme non compatibles avec l’essai ; Les personnes privées de liberté ou sous tutelle ou garde à vue.
- Femmes enceintes ou allaitantes.
- Patients refusant ou incapables de se conformer au suivi médical requis par l’essai pour des raisons géographiques, familiales, sociales ou psychologiques.
Calendrier prévisionnel
Lancement de l’étude : Octobre 2023
Fin estimée ds inclusions : Mai 2026
Nombre de patients à inclure : 96
Coordonnateur de l'étude
Dr Barbara PISTILLI
Gustave Roussy – CLCC Villejuif
Promoteur de l'étude
UNICANCER
Dernière mise à jour le 30 avril 2025