PRODIGE 71 - FFCD 1710 - BEVAMAINT
Essai de phase III comparant le traitement d’entretien par fluoropyrimidine + bévacizumab versus fluoropyrimidine après chimiothérapie d’induction pour un cancer colorectal métastatique
Type d'essai : Académique / Institutionnel
Etat de l'essai : Ouvert
Situation thérapeutique : Tumeur solide ( Métastatique / Rechute )
Etablissement(s) participant(s)
Dr Ludovic EVESQUE
Dr Yves RINALDI
Dr Camille SIBERTIN-BLANC
Dr Bernard PHILIPPE
Dr Jean-François PATEL
Dr Laurent MINEUR
Détails de l'essai
Objectif principal
Temps jusqu’à échec du traitement (TTF).
Objectif(s) secondaire(s)
Toxicité selon NCI-CTC v4.03.
Qualité de vie (selon QLQ-C30).
Survie sans progression (SSP1), selon l’investigateur et selon la relecture centralisé (RECIST V1.1).
Survie sans progression 2 (SSP2), selon l’investigateur et selon la relecture centralisé (RECIST V1.1).
Survie globale (SG).
Résumé / schéma de l'étude
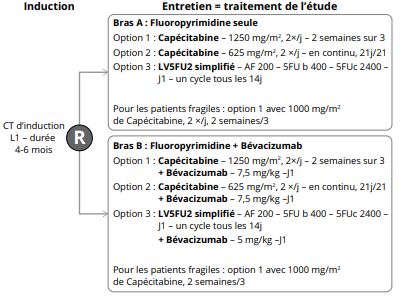
Critère(s) d'inclusion
- Adénocarcinome colorectal métastatique histologiquement prouvé avant la chimiothérapie d’induction.
- Lésion mesurable ou non mesurable avant la chimiothérapie d’induction selon les critères RECIST 1.1.
- Maladie métastatique, non résécable selon la pratique du centre après chimiothérapie d’induction Indice de performance.
- ECOG ≤ 2.
- Contrôle de la maladie (réponse complète, réponse partielle ou maladie stable) après 4 à 6 mois d’une bichimiothérapie (fluoropyrimidine + irinotécan ou oxaliplatine) ou d’une trichimiothérapie (fluoropyrimidine + irinotécan + oxaliplatine) d’induction en première ligne +/- (cétuximab, panitumumab, bévacizumab, ou chimiothérapie IAH).
- Espérance de vie > 3 mois.
- Âge ≥ 18 ans.
- Pas d’intervention chirurgicale majeure dans les 4 semaines avant la randomisation.
- Bilirubinémie totale < 25 μmol/L, ASAT < 3 x LSN, ALAT < 3 x LSN (patient avec métastase hépatique ASAT, ALAT < 5 x LSN), PAL < 2.5 x LSN (patient avec métastase hépatique), PAL < 5 x LSN, TP > 60%.
- Neutrophiles > 1500/mm3, plaquettes > 100 000/mm3, hémoglobine ≥ 9 g/dL
- Clairance de la créatinine > 30 ml/min (MDRD); si la clairance de la créatinine se situe entre 30 et 50 ml/min, voir les RCP pour les adaptations de doses.
- Protéinurie ≤ 2+ (bandelette urinaire) (si plus de 2+, alors la protéinurie des 24h doit ≤ 1g).
- Patient est capable de comprendre, de signer et de dater le consentement éclairé.
- Preuve de la ménopause ou test de grossesse urinaire ou sérique négatif pour les femmes non ménopausées.
- Méthode de contraception médicalement efficace pour les patients en âge de procréer, hommes ou femmes.
- Patient affilié à un système de sécurité sociale.
Critère(s) de non-inclusion
- Infarctus du myocarde, coronaropathie grave ou dysfonction cardiaque grave dans les 6 mois précédent l’inclusion.
- Suivi impossible.
- Patients réséqués de l’ensemble de leurs métastases (R0/R1) après une chimiothérapie d’induction.
- Syndrome Mains-Pieds > 1 avant la chimiothérapie d’entretien.
- Métastase cérébrale ou métastase leptoméningée connue.
- Autre tumeur maligne concomitante ou antérieure, excepté : carcinome in situ traité et en rémission complète pendant plus de 5 ans.
- Hypertension non contrôlée (définie comme une tension artérielle systolique > 140 mmHg et/ou une tension artérielle diastolique > 90 mmHg), ou antécédents d’hypertension ou d’encéphalopathie hypertensive.
- Patiente enceinte ou allaitante.
- Traitement par sorivudine ou analogues (brivudine).
- Traitement par phénytoïne ou analogues.
- Déficit complet et partiel en DPD (Uracilémie ≥ 16 ng/ml).
- Ulcère gastro-duodénal non guéri.
- Toute contre-indications au traitement par bévacizumab ou fluoropyrimidine.
- Fistule ou perforation gastro-intestinale.
- Antécédent ou de saignement gastro-intestinal actif.
- Accident thrombo-embolique et/ou antécédent d’accident thrombo-embolique.
- Insuffisance hépatique sévère.
Calendrier prévisionnel
Lancement de l’étude : Décembre 2019
Fin estimée des inclusions : 2ème trimestre 2026
Nombre de patients à inclure : 400
Information(s) complémentaire(s)
Le sang (plasma) sera prélevé chez tous les patients afin de réaliser des projets de recherche translationnelle (Centre de Ressource Biologique EPIGENETEC, UMR-S 1147, Paris, France, dirigé par le Professeur Pierre Laurent-Puig) sur l’identification de biomarqueurs prédictifs de l’efficacité du traitement (pour plus de détails voir « études ancillaires ») incluant au moins la détection de ADNtc (de base et en cinétique), et la collecte de blocs tumoraux.
Coordonnateur de l'étude
Pr Thomas APARICIO
CHRU Lille
Promoteur de l'étude
CHU Dijon