AcSé pemigatinib
Essai clinique évaluant l’efficacité du pemigatinib pour le traitement des patients atteints de tumeurs solides présentant une altération du gène FGFR
Type d'essai : Académique / Institutionnel
Etat de l'essai : Ouvert
Situation thérapeutique : Tumeur solide ( Métastatique / Rechute , Localement avancée / Non résécable )
Cibles / marqueurs : FGFR
Etablissement(s) participant(s)
Dr Esma SAADA-BOUZID
Dr Cécile VICIER
Dr Bertrand BILLEMONT
Détails de l'essai
Objectif principal
L’objectif de cet essai clinique est d’évaluer l’efficacité du pemigatinib en monothérapie sur la cinétique de croissance tumorale et la réponse tumorale chez des patients atteints d’un cancer récurrent et/ou métastatique (quel que soit le type de cancer à l’exception des cancers du sang et des patients déjà traités par pemigatinib) présentant une altération du FGFR (fusion/réarrangement ou mutation activatrice).
Objectif(s) secondaire(s)
Evaluer l’efficacité en terme :
o Du taux de réponse globale (ORR).
o Du taux de bénéfice clinique (CBR).
o De la durée de réponse (DoR).
o De la survie sans progression (PFS).
o Du temps jusqu’à l’échec du traitement (TTF).
o De la survie globale.
Evaluer la sécurité et la tolérance du pemigatinib.
Evaluer la qualité de vie.
Résumé / schéma de l'étude
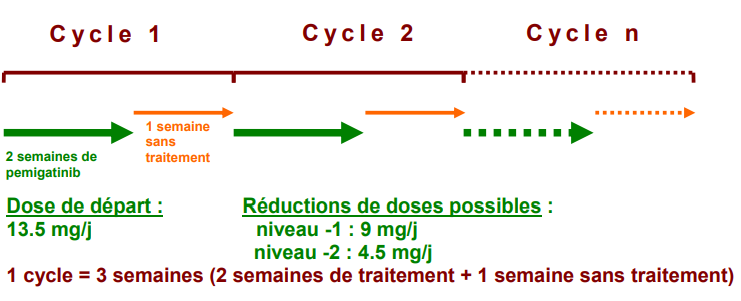
Critère(s) d'inclusion
- Tumeur solide confirmée histologiquement ou cytologiquement.
- Patient atteint d’une maladie localement récurrente non résécable et/ou avancée ou métastatique présentant une fusion/un réarrangement ou une mutation du FGFR1,2,3.
- Âge ≥ 18 ans.
- Statut de performance de l’Eastern Cooperative Oncology Group (ECOG) inférieur ou égal à 2.
- Patient pour lequel il n’existe pas d’alternative thérapeutique appropriée, pour lequel un inhibiteur du FGFR est indiqué par l’institution ou la réunion de concertation pluridisciplinaire régionale, et qui est susceptible de tirer un bénéfice, selon l’évaluation du médecin.
- Espérance de vie estimée à plus de 3 mois.
- Maladie mesurable selon RECIST1.1, quelle que soit la localisation de la maladie. Les lésions tumorales situées dans une zone précédemment irradiée ou dans une zone soumise à une autre thérapie loco-régionale sont considérées comme mesurables si une progression a été clairement démontrée dans la lésion.
- Possibilité de réaliser deux évaluations tumorales avant le traitement, à un intervalle d’au moins quatre semaines et de trois mois au maximum (tomodensitométrie ou IRM, mais avec les mêmes techniques pour les deux), et sans traitement anticancéreux pendant cette période.
- Patient présentant une tendance minimale de 0,1 mm/jour concernant l’augmentation de la cinétique de croissance de la tumeur entre l’examen de pré-traitement et l’examen de référence, telle qu’évaluée par l’investigateur.
- Fonction hématologique adéquate : Neutrophiles > 1,5 x10⁹/L ; plaquettes > 75 x10⁹/L ; hémoglobine > 9,0 g/dL. Les transfusions sont autorisées avec une période d’élimination de 2 semaines avant le début du traitement.
- Fonction hépatique adéquate : ALT et AST < 2,5 x LSN (≤ 5 x LSN en cas de métastases hépatiques) ; bilirubine totale < 1,5 x LSN (< 2,5 x LSN en cas de syndrome de Gilbert ou de métastases hépatiques) ; PAL < 3 x LSN.
- Fonction rénale adéquate : clairance de la créatinine sérique > 30 ml/minute selon la formule de Cockroft-Gault.
- Valeur du phosphate sérique ≤ LSN et valeur du calcium sérique dans la fourchette normale de l’établissement (ou calcium sérique corrigé de l’albumine dans la fourchette normale lorsque l’albumine sérique est en dehors de la fourchette normale).
- Taux de potassium compris dans la fourchette normale de l’établissement ; une supplémentation peut être utilisée pour corriger le taux de potassium au cours du dépistage.
- Les hommes et les femmes en âge de procréer doivent accepter d’utiliser une contraception adéquate pendant toute la durée de leur participation à l’essai et au moins une semaine après avoir terminé le traitement de l’étude. Les hommes doivent également accepter de ne pas faire de don de sperme et les femmes doivent accepter de ne pas faire de don d’ovocytes pendant la période spécifiée.
- Les femmes en âge de procréer doivent subir un test de grossesse négatif dans les 14 jours précédant le début du traitement.
- Le patient est affilié à un système de sécurité sociale.
- Le patient doit avoir signé un formulaire de consentement éclairé avant toute procédure spécifique à l’essai. Lorsque le patient est physiquement incapable de donner son consentement écrit, une personne de confiance de son choix, indépendante de l’investigateur ou du promoteur, peut confirmer en signant le consentement du patient.
Critère(s) de non-inclusion
- Hémopathies malignes.
- Antécédents ou présence d’un ECG anormal qui, de l’avis de l’investigateur, est cliniquement significatif. Un intervalle QTcF de dépistage > 480 ms est exclu.
- Preuve d’une infection active par le virus de l’hépatite B (VHB) ou le virus de l’hépatite C (VHC) (définie par une élévation des transaminases ou une cirrhose ; une infection chronique par le VHB/VHC sans cirrhose ni élévation des transaminases est autorisée).
- Hypersensibilité connue ou réaction sévère au pemigatinib ou aux excipients du pemigatinib (voir la brochure de l’investigateur).
- Patient présentant une maladie et une altération du FGFR couvertes par une indication commercialisée pour tout inhibiteur sélectif du FGFR (par exemple, un cholangiocarcinome avec fusion FGFR2 ou une mutation du FGFR ne sont pas éligibles, alors que la fusion FGFR1 ou 3 est éligible).
- Patient ayant déjà reçu un inhibiteur sélectif du FGFR.
- Patient pouvant être inclus dans une étude de recrutement évaluant un inhibiteur FGFR (y compris le pemigatinib).
- Traitement anticancéreux antérieur, y compris radiothérapie, endocrinothérapie, immunothérapie, chimiothérapie ou autres agents expérimentaux au cours des 4 dernières semaines avant l’inclusion (6 semaines pour les nitrosourées et la mitomycine C). Une période de wash-out d’une semaine est autorisée en cas de radiothérapie palliative d’une maladie ne touchant pas le système nerveux central. Les patients doivent avoir récupéré (≤ Grade 1) des EI des thérapies précédemment administrées ou des traitements locaux avant le début du traitement (à l’exception de l’alopécie, de l’anémie et d’une baisse de la clairance de créatinine).
- Utilisation d’inhibiteurs ou d’inducteurs puissants du CYP3A4 ou d’inducteurs modérés du CYP3A4 dans les 14 jours ou les cinq demi-vies (selon la durée la plus courte) précédant la première dose du médicament à l’étude.
- Toute condition qui, de l’avis de l’investigateur, rendrait indésirable la participation du sujet à l’essai ou compromettrait le respect du protocole.
- L’incapacité ou l’improbabilité du participant à se conformer au calendrier des doses ou aux évaluations médicales et au suivi requis par l’essai pour des raisons géographiques, familiales, sociales ou psychologiques.
- Preuve actuelle d’un trouble cliniquement significatif de la cornée ou de la rétine, confirmée par un examen ophtalmologique.
- Autre tumeur maligne en cours, à l’exception du carcinome in situ du col de l’utérus ayant fait l’objet d’une biopsie conique et du carcinome basocellulaire ou épidermoïde de la peau, traités de manière adéquate. Les survivants du cancer, qui ont subi une thérapie potentiellement curative pour une tumeur maligne antérieure, qui n’ont plus de trace de cette maladie depuis 5 ans ou plus et dont le risque de récidive est jugé négligeable, sont éligibles pour l’essai.
- Antécédents de troubles de l’hémostase du calcium et du phosphate ou de déséquilibre minéral systémique avec calcification ectopique des tissus mous (exception : calcifications couramment observées dans les tissus mous tels que la peau, le rein, le tendon ou les vaisseaux en raison d’une blessure, d’une maladie ou du vieillissement en l’absence de déséquilibre minéral systémique).
- Trouble(s) gastro-intestinal(aux) important(s) susceptible(s) d’interférer avec l’absorption, le métabolisme ou l’excrétion du pemigatinib.
- Infection par le VIH connue, sauf si la charge virale est indétectable.
- Maladie infectieuse chronique active ou actuelle nécessitant un traitement antibiotique, antifongique ou antiviral systémique dans les 2 semaines précédant l’inclusion (les participants présentant des infections chroniques asymptomatiques sous traitement prophylactique sont autorisés).
- Incapacité à avaler et à retenir des médicaments par voie orale.
- Maladie cardiaque cliniquement significative ou non contrôlée, y compris angine instable, infarctus aigu du myocarde dans les 6 mois suivant le jour 1 de l’administration du médicament/traitement à l’étude, insuffisance cardiaque congestive de classe III ou IV de la New York Heart Association et arythmie non contrôlée (les participants munis d’un stimulateur cardiaque ou souffrant de fibrillation auriculaire et dont le rythme cardiaque est bien contrôlé sont autorisés).
- Femmes enceintes ou allaitantes.
- Antécédents d’hypovitaminose D nécessitant des doses supraphysiologiques (par exemple, 50 000 UI/semaine) pour combler la carence.
- Participation à un autre essai thérapeutique dans les 30 jours avant l’inclusion.
- Personnes privées de liberté ou placées sous protection ou tutelle.
Calendrier prévisionnel
Lancement de l’étude : Avril 2025
Fin estimée des inclusions : Juin 2027
Nombre de patients à inclure : 40
Coordonnateur de l'étude
Pr Christophe LE TOURNEAU – Institut Curie – CLCC Paris
Promoteur de l'étude
UNICANCER