PRODIGE 90 - (FFCD 2204) - PREDIR-NEOREC
Dostarlimab néoadjuvant associé à une radiothérapie de courte durée dans le cadre d’une stratégie d’observation et d’attente pour les patients atteints d’un cancer du rectum localement avancé présentant une instabilité des microsatellites ou un déficit de réparation des mésappariements : essai randomisé de phase II
Type d'essai : Académique / Institutionnel
Etat de l'essai : Ouvert
Situation thérapeutique : Tumeur solide ( Néoadjuvant )
Cibles / marqueurs : dMMR / MMRd/MSI-H
Etablissement(s) participant(s)
Détails de l'essai
Objectif principal
Taux d’échec de la stratégie de traitement (TSF) à 24 mois.
Le TSF est défini comme le taux de patients n’ayant pas obtenu de réponse clinique complète (cCR) et ceux ayant obtenu une cCR mais présentant une récidive locale ou métastatique ou une repousse locale dans les 2 ans.
Objectif(s) secondaire(s)
Tolérance (NCI-CTCAE v4.0 évaluant les effets indésirables liés au traitement et non liés au traitement (dostarlimab)).
Taux de préservation des organes à 2 ans.
Taux d’excision locale, d’excision totale du mésorectum ou de création d’une colostomie/iléostomie permanente à 2 ans.
Qualité (R0, complétude du mésorectum) et morbidité (selon les classifications de Clavien, Dindo et Quirke) de l’excision locale ou de l’excision totale du mésorectum ou de la colostomie/iléostomie permanente si nécessaire.
Survie sans maladie (SSM) à 6, 12, 24 et 36 mois.
Survie globale (SG) à 6, 12, 24 et 36 mois.
Changements du score de qualité de vie sur les échelles EORTC QLQ-C30 et QLQ-CR29 (à l’inclusion, à 3, 6, 12 et 24 mois).
Syndrome de résection antérieur LARS (à l’inclusion, à 3, 6, 12 et 24 mois).
Score d’incontinence de WEXNER (à l’inclusion, à 3, 6, 12 et 24 mois).
Echelle de la fonction sexuelle chez l’homme IIEF5, chez la femme FSFI (à l’inclusion, à 3, 6, 12 et 24 mois).
Résumé / schéma de l'étude
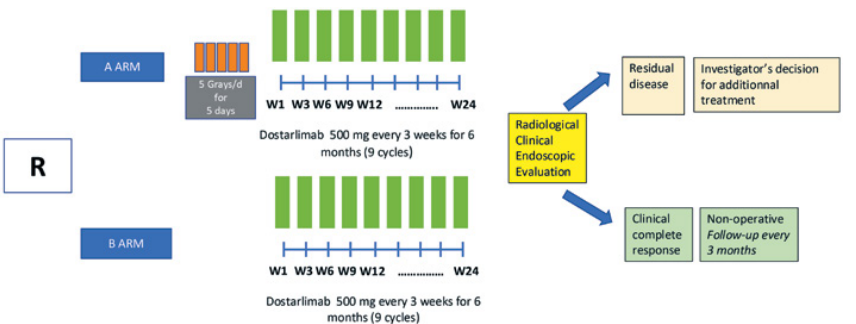
Critère(s) d'inclusion
- Âge ≥ 18 ans.
- Adénocarcinome rectal histologiquement prouvé avec déficit de la réparation des mésappariements (dMMR)/instabilité des microsatellites élevée (MSI-H). Le statut tumoral (dMMR/MSI-H) doit être déterminé par IHC (immunohistochimie) et PCR (réaction en chaîne de la polymérase) ou NGS (séquençage de nouvelle génération).
- Adénocarcinome rectal de stade II ou III et du tiers moyen ou inférieur (diagnostiqué sur la base de critères cliniques et IRM standard).
- Statut de performance de l’OMS 0 ou 1.
- Fonction hépatique adéquate : AST et ALT ≤ 5 x LSN , bilirubine totale ≤ 35 µM/L, albumine ≥ 28 g/L et score de Child-Pugh A (si cirrhose associée).
- Fonction hématologique adéquate (hémoglobine > 9 g/dl, plaquettes > 100 G/L, PNN ≥ 1,5 G/L) et fonction rénale adéquate (clairance de la créatinine ≥ 40ml/min selon la formule MDRD).
- Les femmes en âge de procréer doivent accepter d’utiliser un moyen de contraception pendant l’essai et pendant au moins 4 mois après l’arrêt des traitements expérimentaux. Les hommes ayant des relations sexuelles avec des femmes en âge de procréer doivent accepter d’utiliser un moyen de contraception pendant le traitement et pendant au moins 4 mois après l’arrêt des traitements expérimentaux.
- Capacité du patient à comprendre, signer et dater le formulaire de consentement éclairé avant toute procédure de dépistage spécifique à l’étude.
- Patient affilié à un régime de sécurité sociale.
Critère(s) de non-inclusion
- Adénocarcinome rectal de stade IV et/ou du tiers supérieur (à plus de 10 cm de la marge anale ou sus-péritonéal selon les critères cliniques et IRM standard).
- Patient ayant déjà reçu une immunothérapie, une chimiothérapie ou une radiothérapie pour un cancer du rectum.
- Toxicités persistantes liées au traitement antérieur, de grade supérieur à 1.
- Patient présente une infection active nécessitant un traitement systémique dans la semaine précédant la première dose prévue du traitement à l’étude.
- Contre-indication à la radiothérapie pelvienne.
- Hypersensibilité au dostarlimab ou à l’un de ses excipients.
- Allergie à l’un des composants des cellules ovariennes de hamster chinois.
- Antécédents de maladie auto-immune grave et active mettant en jeu le pronostic vital.
- Antécédents de maladie cardiaque non contrôlée ou symptomatique.
- Maladie pulmonaire interstitielle.
- Métastases du système nerveux central non contrôlées ou méningite carcinomateuse.
- Patient ayant une présence documentée de l’Ag HBs [ou de l’Ag HBc] lors de la sélection ou dans les 3 mois précédant la première administration du traitement protocolaire.
- Le patient a un résultat positif au test de dépistage des anticorps du VHC ou dans les 3 mois précédant la première administration du traitement protocolaire. NOTE : Les patients ayant un résultat positif au test de dépistage des anticorps anti-VHC en raison d’une maladie antérieure résolue ne peuvent être inclus que si un test de confirmation négatif de l’ARN du VHC est obtenu.
- Le patient a un résultat positif au test ARN du VHC lors de la sélection ou dans les 3 mois précédant la première administration du traitement protocolaire. NOTE : Le test ARN du VHC est facultatif et les patients dont le test d’anticorps du VHC est négatif ne sont pas tenus de se soumettre également au test ARN du VHC.
- Infection connue par le VIH.
- Vaccinations (vaccin vivant) dans les 14 jours précédant le début du traitement.
- Immunosuppression, y compris les sujets atteints d’affections nécessitant un traitement par corticostéroïdes systémiques (>10 mg/jour d’équivalent prednisone).
- Maladie auto-immune active nécessitant un traitement systémique (vitiligo exclu) au cours des 2 dernières années ou traitement immunosuppresseur administré dans les 7 jours précédant la première administration du traitement.
- Antécédents de transplantation d’organe.
- Femme enceinte ou allaitante.
- Patient ayant présenté l’une des situations suivantes lors d’une immunothérapie antérieure : tout EI lié à l’immunité (EI) de grade 3 ou plus, événements neurologiques graves liés à l’immunité de tout grade (par ex, syndrome myasthénique/myasthénie grave, encéphalite, syndrome de Guillain-Barré ou myélite transverse), dermatite exfoliative de tout grade (syndrome de Stevens-Johnson, nécrolyse épidermique toxique ou réaction médicamenteuse avec éosinophilie et symptômes systémiques [DRESS]) ou myocardite de tout grade.
- Les anomalies de laboratoire non significatives sur le plan clinique ne sont pas exclusives. Toute maladie évolutive non équilibrée au cours des 6 derniers mois : insuffisance hépatique, insuffisance rénale, insuffisance respiratoire, etc.
- Autre cancer traité au cours des 5 dernières années, à l’exception du carcinome cervical in situ ou du carcinome basocellulaire/spinocellulaire ou d’un cancer du spectre du syndrome de Lynch considéré comme guéri au moment de l’inclusion.
- Intervention chirurgicale majeure dans les 4 semaines précédant l’inclusion.
- Personnes privées de liberté, sous tutelle ou incapables de donner leur consentement.
- Toute condition psychologique, familiale, sociologique ou géographique susceptible d’entraver le respect du protocole de l’étude ou du calendrier de suivi.
Calendrier prévisionnel
Lancement de l’étude : Novembre 2024
Fin estimée des inclusions : Novembre 2026
Nombre de patients à inclure : 68
Promoteur de l'étude
CHU DIJON