GORTEC 2024-03 - IDEAL
Essai de phase II, randomisé, évaluant le traitement d’induction par l’anti PD1 Pucotenlimab et l’EGFRADC MRG003 ou l’induction par l’EGFR-ADC MRG003 seul, suivi par une radiochimiothérapie dans les cancers épidermoïdes localement avancés de la tête et du cou
Type d'essai : Académique / Institutionnel
Etat de l'essai : Ouvert
Situation thérapeutique : Tumeur solide ( Localement avancée / Non résécable )
Etablissement(s) participant(s)
Dr Esma SAADA-BOUZIDD
Pr Sébastien SALAS
Dr Benoit CALDERON
Détails de l'essai
Objectif principal
Evaluer le taux de réponse complète (RC) 12 semaines après la fin de la radio-chimiothérapie , chez les patients CETEC-LA, traités avec une induction par EGFR-ADC MRG003 et un anti PD-1 Pucotenlimab ou traités par une induction par EGFR-ADC MRG003 seul, en utilisant le design Fleming en une étape.
Objectif(s) secondaire(s)
Evaluer le meilleur taux de réponse objective (ORR) à la fin de la phase d’induction par EGFR-ADC MRG003 + anti PD-1 Pucotenlimab ou EGFR-ADC MRG003 seul (21 ± 7 jours après J1 du dernier cycle de traitement d’induction).
Evaluer la qualité de vie dans les 2 bras de traitement.
Evaluer la survie sans progression (SSP) dans les 2 bras de traitement.
Evaluer la survie sans évènement (SSE) dans les 2 bras de traitement.
Evaluer la survie globale dans les 2 bras de traitement, incluant le taux de survie à 24 et 36 mois (SG).
Evaluer la compliance au traitement dans les 2 bras de traitement.
Evaluer les évènements indésirables entre les 2 bras (NCI-CTCAE v5).
Résumé / schéma de l'étude
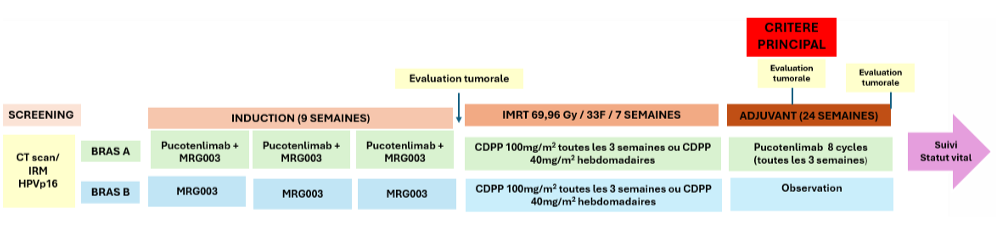
Critère(s) d'inclusion
- Consentement éclairé signé.
- Age ≥18 ans et ≤ 75 ans.
- Score de performance de l’Eastern Cooperative Oncology Group (ECOG) ≤1. (PS).
- Patients porteurs d’un carcinome épidermoïde de la tête et du cou, histologiquement prouvé, non préalablement traité et éligible au traitement par radiochimiothérapie :
- Stade III, IVa ou IVb pour cavité orale, hypopharynx, larynx, oropharynx (p16-) (selon American Joint Committee on Cancer [AJCC]/TNM Staging System, 8th Ed.)
- OU Quelle que soit la consommation de tabac et si carcinome épidermoïde de l’oropharynx p16+ (surexprimé par immunhistochimie) : T3-T4/N1-N3
- OU Si consommation de tabac ≥ 20PA et carcinome épidermoïde de l’oropharynx p16+ : T1T2 / N1-N3 ou T3-T4/N0.
- Pour les patients présentant un carcinome épidermoïde de l’oropharynx, disponibilité du rapport pathologique de détermination du statut HPV (préférentiellement par expression de p16).
- Evaluation tumorale par scanner (CT scan) de la tête et du cou, ou par IRM et basée sur les critères RECIST1.1.
- Patients éligibles à un traitement par cisplatine.
- Pas de perte auditive à l’évaluation clinique ou perte auditive ≤grade 2 (selon NCI-CTCAE v5.0).
- Neuropathie périphérique < grade 2 (selon NCI-CTCAE v5.0).
- Les valeurs de laboratoire du screening doivent répondre aux critères suivants et doivent être obtenues dans les 14 jours précédant la randomisation :
- Neutrophiles polynucléaires > 1,5 x 109/L
- Plaquettes > 100 x 109/L
- Hémoglobinémie > 9,0 g/mL
- ALAT/ASAT < 3 x ULN
- Bilirubine totale < 1,5 x ULN ( sauf syndrome de Gilbert : < 3 mg/dL)
- Clairance de la créatinine ≥ 50 ml/min/1.73m2 (CKD-EPI)
- Disponibilité d’un échantillon tumoral fixé au formol et inclus en paraffine pour détection PD-L1 et expression de la protéine EGFR.
- Aucun traitement antitumoral préalable pour cancer de la tête et du cou, par chimiothérapie, immunothérapie, thérapie ciblée, radiothérapie ou chirurgie.
- Patients ne présentant pas de contre-indications à l’anti-PD-1 Pucotenlimab ou à l’EGFR-ADC MRG003.
- Femmes en âge de procréer doivent avoir un test de grossesse sérique ou urinaire négatif lors du contrôle d’éligibilité et ne doivent pas allaiter. Le test de grossesse doit être effectué dans les 72 heures précédant la randomisation.
- Hommes et femmes (en âge de procréer) sexuellement actifs doivent accepter d’utiliser des méthodes contraceptives hautement efficaces depuis la signature de l’ICF jusqu’à au moins 6 mois après la fin du traitement (si le dernier traitement reçu est MRG003 ou pucotenlimab) ou 7 mois après la fin de traitement (si le dernier traitement reçu est le cisplatine).
- Patients disposant d’une couverture d’assurance sociale.
Critère(s) de non-inclusion
- Tumeur primitive du nasopharynx, des sinus paranasaux, de la cavité nasale, des glandes salivaires, de la thyroïde ou de la glande parathyroïde, de la peau ou carcinome épidermoïde de primitif inconnu ou cancer non épidermoïde (ex mélanome muqueux).
- Maladie métastatique (stade IVc selon AJCC/TNM, 8th Ed.).
- Patients ayant reçu une thérapie précédente par anti-PD-1, anti PD-L1 , anti PD-L2, anti CD137 ou anticorps anti-CTLA-4 (ou tout autre anticorps ou médicament ciblant spécifiquement les voies de co-stimulation ou de point de contrôle des lymphocytes T).
- Traitement pour une autre pathologie avec un produit ou un dispositif médical expérimental dans les 4 semaines de la première dose du traitement à l’étude.
- Antécédents de tumeur maligne au cours des 3 années précédentes, à l’exception des cancers de la peau non-mélanomes complètement réséqués en dehors de la région de la tête et du cou ou des cancers du sein de stade I complètement réséqués, ou des carcinomes in situ non musculaires invasifs de la vessie, du col de l’utérus, de l’utérus et/ou de la prostate (Gleason6) complètement réséqués ou carcinome épidermoïde T1a de l’œsophage ou du rectum/anus.
- Patients atteints d’une maladie cardiovasculaire cliniquement significative (i.e, active) : accident vasculaire cérébral (< 6 mois avant l’inclusion), infarctus du myocarde (< 6 mois avant l’inclusion), angor instable, insuffisance cardiaque congestive (≥ Classification de la New York Heart Association Classe II), ou arythmie cardiaque grave nécessitant un traitement médicamenteux, ou connaissance d’une fraction d’éjection ventriculaire gauche réduite et persistante < 50%.
- Patients avec test positif pur le virus de l’immunodéficience humaine (HIV) ou syndrome d’immunodéficience acquise connu (SIDA).
- Patients avec test positif pour l’antigène de surface du virus de l’hépatite B (AgHBV) ou acide ribonucléique du virus de l’hépatite C (ARN du VHC) indiquant une infection active ou chronique. Présence d’une autre pathologie hépatique, incluant les maladies hépatiques auto-immunes chroniques, la cirrhose biliaire ou la cholangite sclérosante.
- Autres infections actives (virales et/ou bactériennes et/ou mycotiques) nécessitant un traitement systémique le jour précédent la randomisation.
- Vaccins vivants atténués dans les 4 semaines précédant la première administration du traitement à l’étude. L’utilisation du vaccin inactivé contre la grippe saisonnière ou d’un vaccin COVID-19 approuvé ne contenant pas de virus vivants sont autorisés.
- Perte de poids > 10% durant les 4 semaines précédants la randomisation (sauf si des mesures adéquates sont mises en place pour assurer un support nutritionnel).
- Saignement gastro-intestinal actif, ou tout autre saignement non contrôlé nécessitant plus de 2 transfusions sanguines ou 4 culots globulaires dans les 4 semaines précédant la randomisation.
- Condition médicale connue ou sous-jacente (ex : condition médicale associée à une diarrhée ou diverticulite aiguë), qui selon l’investigateur, rendrait l’administration du traitement à l’étude dangereuse pour le patient ou rendrait plus difficile l’interprétation des toxicités ou la détermination des évènements indésirables.
- Maladie inflammatoire active non contrôlée (incluant polyarthrite rhumatoïde, lupus erythémateux, syndrome de Sjögren, psoriasis sévère étendu and autre maladie autoimmune) nécessitant un traitement par anti-TNF. Les sujets atteints de vitiligo, de diabète de type I, d’hypothyroïdie résiduelle due à une maladie auto-immune nécessitant uniquement un remplacement hormonal, un psoriasis ne nécessitant pas de traitement systémique ou des conditions qui ne devraient pas se reproduire en l’absence de déclencheur externe sont éligibles.
- Patients présentant une affection nécessitant une administration systémique chronique de corticostéroïdes (> 10 mg d’équivalents prednisone par jour) ou d’autres médicaments immunosuppresseurs dans les 14 jours avant la première administration du médicament à l’étude. Les stéroïdes inhalés ou topiques et les doses de remplacement surrénalien > 10 mg par jour d’équivalents prednisone sont autorisés en l’absence de maladie auto-immune active.
- Antécédents de convulsions incontrôlées, de troubles du système nerveux central ou de handicap psychiatrique jugé par l’investigateur comme cliniquement significatif, excluant le consentement éclairé ou interférant avec la compliance.
- Maladie(s) concomitante(s) grave(s), y compris des troubles psychiatriques qui limiteraient le respect des exigences de l’étude.
- Antécédents de greffe d’organe, y compris greffe de cellules souches périphériques allogéniques ou de moelle osseuse. Les patients ayant reçu une greffe autologue de cellules souches hématopoïétiques (HSTC) il y a ≥ 5 ans et ayant une fonction médullaire normale (non dépendante des transfusions) pourraient être inclus.
- Patient sous tutelle, curatelle ou privé de liberté.
Calendrier prévisionnel
Lancement de l’étude : Octobre 2025
Fin estimée des inclusions : Octobre 2026
Nombre de patients à inclure : 106
Coordonnateur de l'étude
Pr Jean BOURHIS – Groupe Oncologie Radiotherapie Tete et Cou (GORTEC)
Promoteur de l'étude
Groupe Oncologie Radiotherapie Tete et Cou (GORTEC)